Glide Methodology
Glide uses a hierarchical series of filters to search for possible locations of the ligand in the active-site region of the receptor. The shape and properties of the receptor are represented on a grid by several different sets of fields that provide progressively more accurate scoring of the ligand poses.
Stage 1
Conformational flexibility is handled in Glide by an extensive conformational search, augmented by a heuristic screen that rapidly eliminates unsuitable conformations, such as conformations that have long-range internal hydrogen bonds.
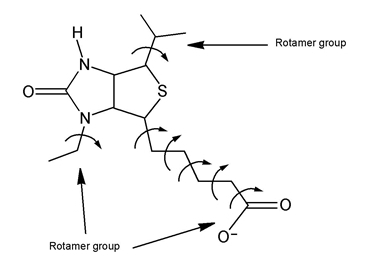
Figure 1:Definition of core and rotamer groups.
As illustrated in Figure 1, each ligand is divided into a core region and some number of rotamer groups. Each rotamer group is attached to the core by a rotatable bond, but does not contain additional rotatable bonds. The core is what remains when each terminus of the ligand is severed at the “last” rotatable bond. Carbon and nitrogen end groups terminated with hydrogen (—CH3, —NH2, —NH3+) are not considered rotatable because their conformational variation is of little significance. In Figure 1, the four central torsions are part of the core, and the methyl groups are not considered rotatable.
During conformation generation, each core region is represented by a set of core conformations, the number of which depends on the number of rotatable bonds, conformationally labile 5- and 6-membered rings, and asymmetric pyramidal trigonal nitrogen centers in the core. This set typically contains fewer than 500 core conformations, even for quite large and flexible ligands, and far fewer for more rigid ligands. Every rotamer state for each rotamer group attached to the core is enumerated. The core plus all possible rotamer-group conformations is docked as a single object. Glide can also dock sets of pre-computed conformations. However, Glide offers its greatest value when conformations are generated internally.
For each core conformation (or for rigid docking, each ligand), an exhaustive search of possible locations and orientations is performed over the active site of the protein. The search begins with the selection of “site points” on an equally spaced 2 Å grid that covers the active-site region (Stage 1 in Figure 2). This selection is made as follows. Distances from the site point to the receptor surface are evaluated at a series of pre-specified directions and sorted into distance ranges (“bins”) of width 1 Å. Likewise, distances from the ligand center (the midpoint of the two most widely separated atoms) to the ligand surface are sorted into bins of width 1 Å. For a given site point, the distance ranges from the site point to the receptor are compared with those from the ligand center to the ligand surface. Glide positions the ligand center at the site point if there is a good enough match, but skips over the site point if there is not.
Stage 2
The second stage of the hierarchy begins by examining the placement of atoms that lie within a specified distance of the line drawn between the most widely separated atoms (the ligand diameter). This is done for a pre-specified selection of possible orientations of the ligand diameter (Step 2a). If there are too many steric clashes with the receptor, the orientation is skipped.
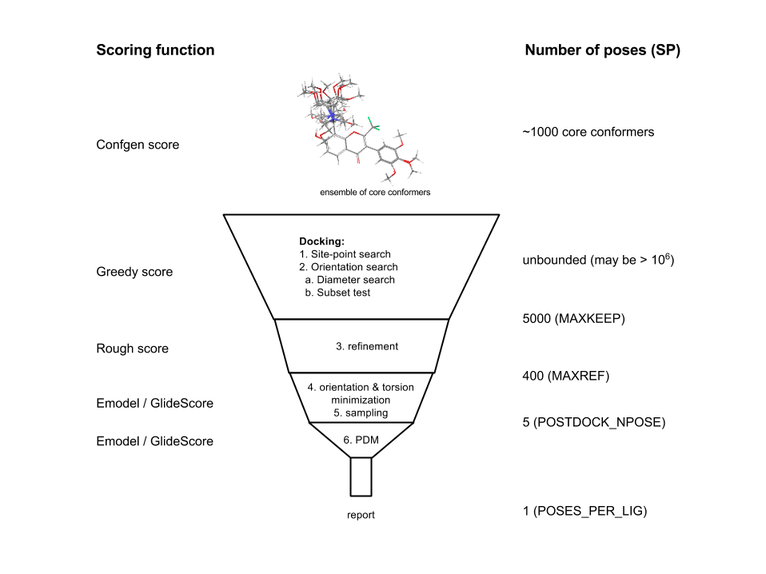
Figure 2:The Glide docking hierarchy.
Next (Step 2b), rotation about the ligand diameter is considered, and the interactions of a subset consisting of all atoms capable of making hydrogen bonds or ligand-metal interactions with the receptor are scored (subset test). If this score is good enough, all interactions with the receptor are scored (Step 2c).
The scoring in these three tests is carried out using Schrödinger’s discretized version of the ChemScore empirical scoring function [1]. Much as for ChemScore itself, this algorithm recognizes favorable hydrophobic, hydrogen-bonding, and metal-ligation interactions, and penalizes steric clashes. This stage is called “greedy scoring,” because the actual score for each atom depends not only on its position relative to the receptor but also on the best possible score it could get by moving ±1 Å in x, y, or z. This is done to mute the sting of the large 2 Å jumps in the site-point/ligand-center positions. The final step in Stage 2 is to re-score the top greedy-scoring poses via a “refinement” procedure (Step 2d), in which the ligand as a whole is allowed to move rigidly by ±1 Å in the Cartesian directions.
Only a small number of the best refined poses (typically 100-400) is passed on to the third stage in the hierarchy—energy minimization on the pre-computed OPLS van der Waals and electrostatic grids for the receptor. The energy minimization typically begins on a set of van der Waals and electrostatic grids that have been “smoothed” to reduce the large energy and gradient terms that result from too-close interatomic contacts. It finishes on the full-scale OPLS nonbonded energy surface (“annealing”). This energy minimization consists only of rigid-body translations and rotations when external conformations are docked. When conformations are generated internally, however, the optimization also includes torsional motion about the core and end-group rotatable bonds. Unless otherwise specified, a small number of the top-ranked poses are then subjected to a sampling procedure in which alternative local-minima core and rotamer-group torsion angles are examined to try to improve the energy score.
GlideScore
The units of GlideScore are kcal/mol. Both SP and XP GlideScore have been calibrated to reproduce certain experimental binding affinities. The individual terms (Emodel, Ecoul, and so on) in the GlideScore formula are parametrized in this calibration: these terms should therefore not be interpreted as quantitative calculations of energies.
GlideScore is based on ChemScore, but includes a steric-clash term, adds other rewards and penalties such as buried polar terms (devised by Schrödinger to penalize electrostatic mismatches), amide twist penalties, hydrophobic enclosure terms, and excluded volume penalties, and has modifications to other terms:
GScore = 0.05*vdW + 0.15*Coul + Lipo + Hbond + Metal + Rewards + RotB + Site
The components of the GlideScore (GScore) are described in Table 1.
The choice of best-docked structure for each ligand is made using a model energy score (Emodel) that combines the energy grid score, the binding affinity predicted by GlideScore, and (for flexible docking) the internal strain energy for the model potential used to direct the conformational-search algorithm.
Glide also computes a specially constructed Coulomb-van der Waals interaction-energy score (CvdW) that is formulated to avoid overly rewarding charge-charge interactions at the expense of charge-dipole and dipole-dipole interactions. This score is intended to be more suitable for comparing the binding affinities of different ligands than is the “raw” Coulomb-van der Waals interaction energy. In addition to the GlideScore, a “docking score” is reported, which is the GlideScore supplemented by Epik state penalties, if used, and strain corrections, if used.
This hierarchical search gives Glide exceptionally high accuracy in predicting the binding mode of the ligand. At the same time, the computational cost is dramatically reduced compared to what would be required for a complete systematic search. The key to this reduction is that the algorithm allows the rotamer groups to be optimized one at a time for a given core conformation and location of the ligand. For example, if there are five rotamer groups and each has three rotamer states, the total number of conformers in the ensemble based on this core conformation/location is 35 = 243. However, if the rotamer groups are optimized one at a time, the number of conformational combinations is only 3×5 = 15, for a savings of about a factor of 15 in computational effort. While many other time-saving algorithms in Glide contribute to its performance advantages, this fundamental qualitative feature allows large libraries to be screened at an affordable computational cost.