Symmetrizing a Molecule
By default, Jaguar takes advantage of molecular symmetry1 whenever possible, in order to save CPU time. Both Abelian and non-Abelian point groups are recognized. Generally, you should symmetrize the geometry if you plan to use symmetry in the calculation itself. Otherwise, the input coordinates may not be accurate enough for the desired symmetry to be recognized.
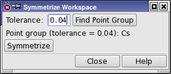
You can symmetrize the molecule by choosing Edit → Symmetrize Workspace. The Symmetrize Workspace panel opens.
The point group symmetry is determined as follows. After the molecule is translated so that the center of mass is at the origin of the coordinate system and rotated so that the principal axes of inertia are aligned on the coordinate axes, symmetry operations (reflections, rotations, and inversions) are applied to determine the point group of the molecule.
When Maestro checks whether a symmetry operation produces an equivalent structure, the coordinates of the two structures only have to be the same to within a prescribed tolerance, that is, each pair of symmetry-related atoms is within a distance specified by the tolerance. The value of the tolerance can be specified in the Tolerance text box, and is 0.04 Å by default. This value ensures that the highest symmetry is found in most cases. By changing the value and clicking the Find Point Group button, you can determine whether there is a lower (or higher) symmetry point group that approximately describes the structure, and use that group to symmetrize the molecule instead of the default.
The tolerance is also used when the molecule is symmetrized. After translation and rotation, the coordinates of the atoms are adjusted to reflect the symmetry group accurately. The maximum displacement permitted is the tolerance specified. A large tolerance yields the highest symmetry, but may cause the coordinates to be changed significantly. A small tolerance may yield a lower symmetry, but results in smaller coordinate changes. The main Jaguar programs use a small tolerance (1.0 x 10−6 bohr), which should result in molecular energy changes of 1 microHartree or less.
You can turn the use of symmetry off2 in the Molecule tab. For methods such as LMP2 and ZORA, and for some properties such as IR intensities or hyperpolarizabilities, symmetry is not implemented and is disabled automatically for the job.
If you are comparing calculations from geometries that differ only slightly, you must use caution when symmetrizing coordinates. For example, a small symmetry-breaking change can be removed if its magnitude is smaller than the tolerance you have set, which establishes what changes are acceptable. In this case, you should inspect the symmetrized coordinates in the Edit Job dialog box to ensure that symmetrizing had the desired effect and did not discard any important information about the molecular geometry.