Ab initio Quantum Chemical Calculation of pKa Values
The calculation of the pKa of a molecule in aqueous solution can be represented as a thermodynamic cycle:
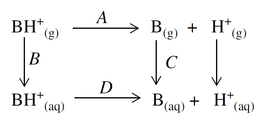
The strategy in our pKa module is to calculate parts A, B, and C of the above cycle, whereupon the actual pKa, which is related to D by

can be obtained by summing the free energy changes for these three components and the experimental value of −259.5 kcal/mol for the solvation free energy change of a proton.
Segment A is the gas phase reaction:
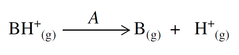
The gas phase free energy difference between the protonated and deprotonated states can be computed via the usual relations

Evaluation of this expression requires the following quantum chemical calculations:
| 1. | Geometry optimization of the protonated and deprotonated species. We assume here that there is only a single, well-defined conformational minimum and that a good initial guess, obtained, for example, from molecular mechanics or semiempirical quantum chemistry, is available. Density functional theory, particularly the hybrid methods that include Hartree-Fock exchange, have been shown to provide good quality geometries; we compute geometries at the B3LYP/6-31G* level of theory with a PCM implicit solvation model. |
| 2. | Accurate single-point energies at each optimized geometry must be evaluated. These single-point calculations are carried out at a significantly higher level of theory than the geometry optimization, but since only one energy is required, the overall cost of this step is less than that for geometry optimization. In recent publications, and in our own extensive unpublished work, the B3LYP method with large basis sets has been shown to yield excellent gas phase energetics for deprotonation reactions, with errors typically in the 1-3 kcal/mol range. We use the cc-pVTZ(-f)+ basis set of Dunning and coworkers in the present methodology. The residual errors in the DFT calculations appear to be relatively constant for a given functional group as the substituents are altered, and hence can be largely removed by the empirical corrections. |
| 3. | The solvation free energy of the protonated and deprotonated species must be computed. |