Spin States Panel
Calculate high-spin and low-spin states of transition metal complexes, exploring some or all of the possible states produced by Jaguar's transition metal initial guess code. Calculations can also be done on organic molecules and on complexes with more than one transition metal.
To open this panel, click the Tasks button and browse to Quantum Mechanics → Spin States.
The following licenses are required to use this panel: MS Maestro, Jaguar
- Using
- Features
Using the Spin States Panel
This panel enables you to generate spin states for transition metal complexes. It is primarily intended for complexes with a single metal, where the possible occupations of the d orbitals can be explored. You can search for the ground state, a specific spin state, or all spin states, either for the default initial guess of the orbital occupations, or for all occupations generated from the open d shell. You can obtain adiabatic excitation energies by optimizing the geometry of each state; otherwise the energy differences are the vertical excitation energies.
It is also possible to generate spin states for organic molecules. In this case, the unpaired electrons go into the lowest unoccupied molecular orbitals after the paired electrons have been assigned to orbitals.
For mononuclear transition metal complexes, you must assign formal charges to the metal center and the ligands so that the ligands are represented as closed-shell molecules. Assigning atomic formal charges helps Jaguar to construct initial guesses for metal complexes that sample different arrangements of electrons in the d orbitals of the metal center. The spin states facility was designed to sample only the open-shell character of the d orbitals, and requires the ligands to be closed shell. The formal charge on the entire complex is ignored: only the atomic formal charges are taken into account. The charge on the complex can of course be constructed from the atomic formal charges.
To prepare a complex with the correct charges, you can use the tools in the 3D Builder panel.
To assign formal charges to a complex:
-
Ensure that all metal-ligand bonds are of zero order.
You can use the Decrement bond order button in the 3D Builder panel to do this.

-
Display formal charges on the atoms.
Choose Formal Charge from the Apply Labels button menu in the Style toolbox. You can click this button again to reapply charge labels.
-
Set the formal charges on the ligand atoms and the metal atom.
You can use the Increment formal charge and Decrement formal charge buttons in the 3D Builder panel to do this.


You should consider the bonding as ionic, and make the ligands negative and the metal positive. For example, halogens should have a charge of −1, and the two nitrogens in a porphyrin that bond to the metal should have a charge of −1. The nitrosyl ligand would generally be treated as NO−, but may be treated as NO+ when studying solvation effects. The overall charge of the complex is reported in the status bar.
These classical atomic formal charge and closed shell ligand assignments are used simply to provide flexibility in the SCF initial guesses. They have no relation to the final converged wave functions, which can have any distribution of atomic charges and spins.
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
Spin States Panel Features
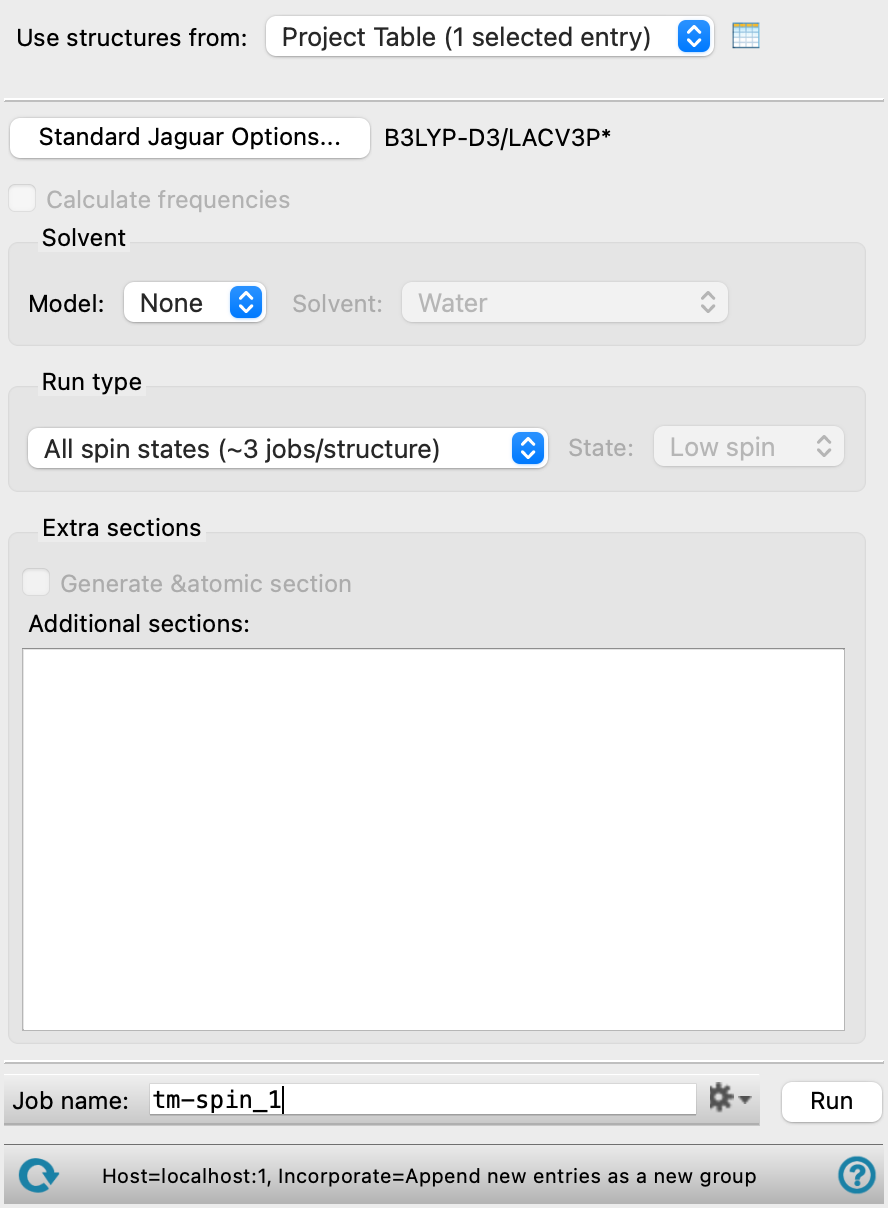
- Use structures from option menu
-
Choose the structure source for the spin states calculation.
- Project Table (n selected entries)—Use the entries that are currently selected in the Project Table or Entry List. The number of entries selected is shown on the menu item.
- Workspace (n included entries)—Use the entries that are currently included in the Workspace, treated as separate structures. The number of entries in the Workspace is shown on the menu item.
- File—Use the specified file. When this option is selected, the File name text box and Browse button are displayed.
- Project Table (n selected entries)—Use the entries that are currently selected in the Project Table or Entry List. The number of entries selected is shown on the menu item.
- Open Project Table button
-
Open the Project Table panel, so you can

- File name text box and Browse button
-
Enter the file name in this text box, or click Browse and navigate to the file. The name of the file you selected is displayed in the text box.
- Options button
-
Set Jaguar options for the spin states calculations. Opens the Jaguar Options - Spin States Dialog Box. This dialog box also allows you to specify additional Jaguar keywords. The solvent (if any), level of theory, and basis set are shown to the right of the button.
-
If you minimize the geometry for the spin states, the energy differences between the states are the adiabatic excitation energies. By default, only an energy calculation is done, and the energy differences are the vertical excitation energies.
- Calculate frequencies option
-
Calculate the vibrational frequencies for each spin state. This option substantially increases the time taken for the job. It is only available if you choose to minimize the geometry when setting Jaguar options.
- Solvent section
-
Choose a solvent model from the Model option menu. The choices are None, which requests a calculation in vacuum, and PBF, which uses the Poisson-Boltzmann solver with a continuum solvent. If you select the latter option, you can then choose the solvent from the Solvent option menu.
- Run type section
-
Choose the states for which you want to perform calculations in this section. The first option menu allows you to choose whether to do calculations for just the selected spin state or all spin states, and for the default initial guess of the d-orbital occupations, or all possible initial guesses. If you are running the calculation on an organic molecule or a complex with more than one transition metal, the initial guess options are not available.
If you choose to select the spin state, the State option menu is activated, and you can choose to optimize the low-spin state (singlet or doublet) or the high spin state (triplet or quartet for organics or systems with more than one metal, highest allowed spin state for single-metal complexes). For octahedral complexes with a single 3d transition metal, a third option, ground, is available, which uses a spin multiplicity value that has been predicted by a physical model to be that of the ground electronic state. In the case of spin-crossover systems, choosing ground selects all of the relevant quasi-degenerate spin multiplicities.
- Extra sections section
-
Specify extra sections for the Jaguar input file. You can add the text for these sections in the text box. The Generate &atomic section option adds an atomic section with formal charge (formal) and multiplicity (multip) for the metal atom. You could, for example include zvar and coord sections to do a coordinate scan.
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Spin States - Job Settings Dialog Box, where you can make settings for running the job.
- Status bar
-
The status bar displays information about the current job settings and status for the panel. The settings includes the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
Use the Reset button
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel. The status bar also contains the Help button
 , which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a
, which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a  button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.
button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.