Jaguar pKa Calculations
pKa calculations consist of a series of calculations on the protonated form and on the deprotonated form of the target molecule, followed by an empirical correction.
Note: The pKa module requires a special license in addition to the regular Jaguar license. When you request a Jaguar license, you should explicitly indicate in your license request that you want to run pKa calculations.
To set up and submit pKa calculations from Maestro, select the entries in the Project Table or Entry List for which you want to calculate the pKa, then choose click the Tasks button and browse to Quantum Mechanics → pKa. The Jaguar panel opens with the Input tab displayed.
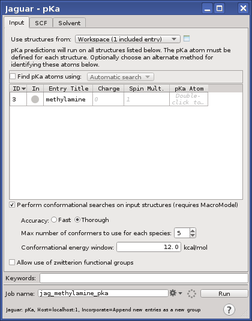
For each structure, the atom or atoms whose pKa you want to calculate can be specified in the pKa Atom cell in the Input tab. Click in this cell, then pick the atoms in the Workspace, or enter the atom names separated by commas. If the structure is not already in the Workspace, it is placed in the Workspace (as the only structure). If you pick more than one atom, calculations will be run for each atom that you pick, and thus you can calculate the pKa for each site in a single molecule that has multiple protonation or deprotonation sites
A pKa atom should be the acidic hydrogen atom in an acid, or the basic atom in a base. You can use the conjugate acid or base as input, but you should choose the hydrogen for a conjugate acid such as an ammonium ion or the heavy atom for the conjugate base such as a carboxylate.
Note: Molecules in which the pKa atom is involved an intramolecular hydrogen bond are likely to give erroneous results, so you should not run calculations on these molecules. You should consider using Epik for these systems.
For microscopic pKa calculations, in which only a single conformation of the molecule is used, it is critical to choose the lowest energy conformer. To perform a conformational search on the protonated and the deprotonated species with MacroModel before the pKa calculation is performed, select Perform conformational searches on input structures, and ensure that Max number of conformers to use for each species is set to 1. You can also choose an option for the accuracy of the conformational search: Fast or Thorough. The lowest-energy conformer is used for each species. This option requires a MacroModel license.
For macroscopic pKa calculations, set Max number of conformers to use for each species to a number greater than 1 to generate the required conformations. You can also set a threshold on the conformational energy in the Conformational energy window text box to filter out high-energy conformers. See Conformational Flexibility in Jaguar pKa Calculations for more information on the macroscopic pKa calculations. This is the default calculation type.
For amino acid zwitterions, the conformational search is skipped, even if this option is selected. You can also disable the specific zwitterion parametrization by deselecting Allow use of zwitterion functional groups.
When setting up a pKa job for a zwitterion, make sure that the structure is set up such that a zwitterion species is present during the calculation. For example, if you are setting up an amino acid as base calculation with the amine group to be protonated, make sure that the carboxylic acid group is deprotonated with a charge of −1 on the oxygen, so that protonation of the amine results in a zwitterion species. You should not, however, create formal charges on a structure for which there is a resonance structure with no formal charges, such as a lactam. If you do, the formal charges and bond orders are reassigned to remove the formal charges before identifying the functional group.
The other tabs available for pKa calculations are the SCF tab, in which you can make settings to control the SCF convergence, and the Solvent tab, in which you can choose the solvent for the pKa calculation. If you want to calculate the pKa for a CH acid or an NH acid in DMSO, for which parameters are available, you must choose DMSO as the solvent; otherwise the solvent should be water. These are the only solvent choices for pKa calculations. You can also select Use solvation for geometry optimization step to optimize the geometries in the chosen solvent; otherwise they are optimized in the gas phase. Optimization in the solvent is more expensive but produces better results.
When you have finished setting up the job, click Run to start the job or click the Settings button  to make job settings in the Job Settings Dialog Box, then click Run to submit the job. See Running Jaguar Jobs for information on job settings. You can distribute a job for multiple molecules across multiple processors.
to make job settings in the Job Settings Dialog Box, then click Run to submit the job. See Running Jaguar Jobs for information on job settings. You can distribute a job for multiple molecules across multiple processors.
The pKa values for each atom are added to the structure in the Maestro output file as values of an atom property (r_j_pKa_water). The output structure is the structure obtained from a geometry optimization of the input structure. The pKa value for the first pKa atom is also added to this structure as an entry property. When a pKa job is incorporated, the property is listed in the Project Table, and the pKa atoms are labeled with the pKa values.