Introduction to Macro-pKa
A pKa calculation of a molecule requires the knowledge of at least one site at which protonation/deprotonation happens. For simple molecules with one such site and no possibility for tautomerism, it is relatively straightforward to manually identify where the transition should occur. For more complex molecules such as
-
Molecules with several protonatable/deprotonatable functional groups whose protonation/deprotonation may not occur in clearly separated pH regions
-
Molecules with many protonatable/deprotonatable groups and potentially multiple protonation/deprotonation states
-
Molecules prone to tautomerism
Multiple tautomeric states and protonation/deprontonation sites might need to be considered at a given pH. For these cases, the risk of selecting an incorrect site, and consequently working with unphysical protonation states becomes too high for a manual selection process.
The Macro-pKa prediction module uses a physics-based method to automatically generate tautomers and identify relevant protonation/deprotonation sites. Empirically corrected micro-pKas for these structures are calculated, and combined to give a macro-pKa which can be directly compared to experimentally measured pKas.
Note: The Macro-pKa module requires a Macro-pKa license in addition to the Jaguar license.
The Macro-pKa workflow
Here we describe the general workflow for how the Macro-pKa module calculates the macro-pKa of a given charge transition. For more information about how charge transitions are defined in the Macro-pKa module, see Defining Charge Transitions.
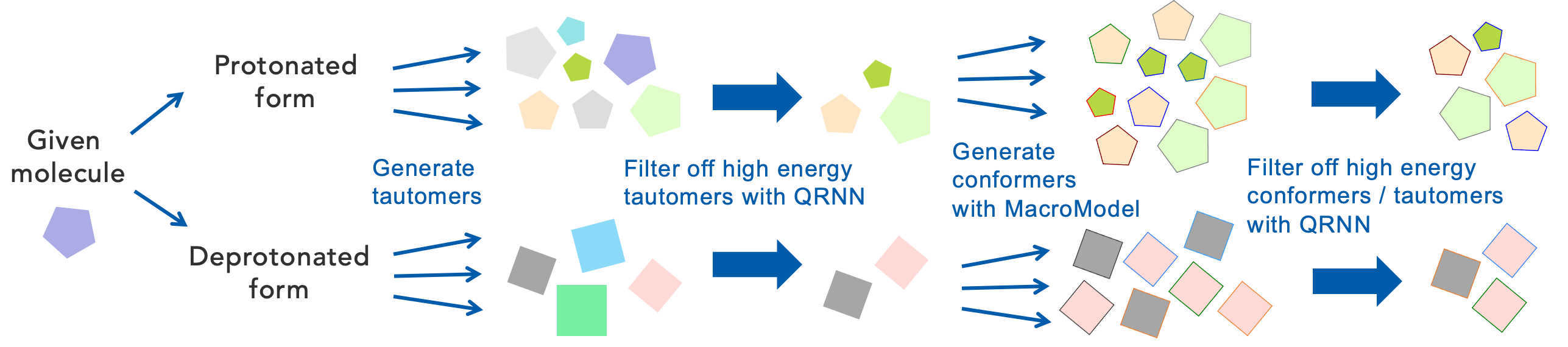
Structure Generation
-
Physics-based automatic generation of tautomers
-
Use machine learning (QRNN) methods to exclude high energy tautomers. If structures contain elements not supported by QRNN, use semi-empirical (PM7) methods instead.
-
Use MacroModel to generate conformers for each resulting tautomer
-
Use machine learning (QRNN) methods to exclude high energy tautomers and conformers. If structures contain elements not supported by QRNN, use semi-empirical (PM7) methods followed by a finer DFT scoring instead.
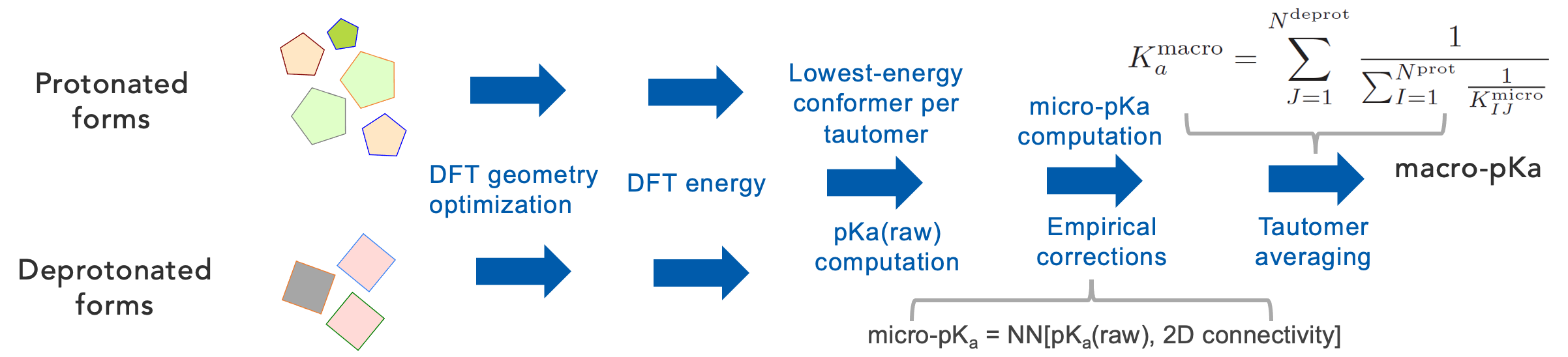
Computing macro-pKa
1. For the resulting tautomers and conformers from the structure generation, calculate raw micro-pKas using ab initio quantum chemical methods
2. Convert raw micro-pKas into micro-pKas using atomic graph convolutional neural networks (GCNN)
3. Statistical averaging over tautomers to obtain the final macro-pKa
A note on terminology
The following terms are used throughout the discussion of the Macro-pKa, and are listed here to clarify their very specific meanings.
-
micro-pKa— The negative log of the equilibrium constant that describes the protonation/deprotonation equilibrium of a molecule in a single tautomeric state. (More precisely, the micro-pKa refers to pKa of the lowest-energy conformer for a given tautomer).
-
macro-pKa— The negative log of the equilibrium constant that describes the protonation/deprotonation equilibrium of multiple tautomers. This value is directly comparable with experimentally measured pKa. The micro-pKa is equivalent to the macro-pKafor molecules with only one possible protonation and deprotonation state.
-
Macro-pKa and Jaguar pKa (Micro-pKa)— Names of the prediction modules used to compute pKa. Jaguar pKa cannot directly compute macro-pKas, while Macro-pKa computes both micro-pKas and macro-pKas.
Resources
To run Macro-pKa in Maestro, see pKa Panel.
To run Macro-pKa on the command line, see Running Macro-pKa from the Command Line.
See pKa Prediction with Macro-pKa for a tutorial.
See the following for more details about each stage of a Macro-pKa calculation: