Amorphous Properties Panel
Calculate properties for molecules in an amorphous structure, taking into account the immediate environment of each molecule. The properties available for calculation are NMR shieldings, and singlet and triplet excitation energies by TDDFT with the S1-T1 gap also reported.
To open this panel, click the Tasks button and browse to Quantum Mechanics → Amorphous Properties.
The following licenses are required to use this panel: MS Maestro, Jaguar
- Using
- Features
- Additional Resources
Using the Amorphous Properties Panel
The properties for the amorphous structure are calculated using Jaguar. For each molecule, the structure is trimmed down to the active molecule and its neighbors, which are defined by a distance cutoff (set in the Nearest neighbor distance text box). This group of molecules is referred to as a "cluster". Periodic boundary conditions are used on the input structure to generate structures in the adjacent unit cell if the neighbors of any active molecule span the cell boundaries.
The input structure can be a pure substance or a mixture of different molecules, but it must be a single Maestro entry. If importing the input from a file, the file must be a Maestro or Desmond CMS file. You can create an amorphous structure with the Disordered System Builder.
By default, a smaller basis set is used for the neighbors than for the active molecule, as they mainly constitute a perturbation to the electronic environment of the target. The neighbors can also be represented by point charges, taken either from a Maestro charge property, or from a Jaguar ESP charge calculation. Using point charges speeds up the calculation considerably, but with a potential loss of accuracy.
You can change the basis sets for the active molecule and the spectators (neighbors) and also choose the DFT method for the calculations. For NMR calculations, you can optimize the bonds to hydrogen atoms or all bonds, or not do any optimization. Only the active molecule is optimized, the spectators are fixed at their input geometries. TDDFT calculations of electronic excitations are done at the existing geometries.
You can run calculations on all molecules, a random selection of molecules, or a selection of molecules, by selecting one of the Molecule subset choices. If you want to select individual molecules, you can do so by listing the molecule numbers in the Molecules text box. Two ways you can identify the molecule number in the Workspace are:
-
Pause the pointer over an atom. The molecule number is displayed in the status bar of the Workspace (atom number first, then molecule number).
-
Label the molecules with the molecule number, by choosing Molecule number from the Apply Label s button menu in the Style toolbox.

As the calculations can take some time, it is recommended that you distribute them over multiple processors, which you can do in the Job Settings dialog box.
The NMR shieldings are written to a text file, jobname.txt. This file has six tab-separated columns: the atom label (element and atom number), the output file, the shielding, and the three diagonal elements of the shielding tensor. The atoms are sorted by label, so that all the results for a given atom in different instances of each molecule are listed together for comparison. The output file naming convention is Cluster_molecule-number.out.
The TDDFT properties (excitation energies and oscillator strengths) are stored as Maestro properties in the output structure file. Note that as the excited states cannot be localized to the active molecule when the spectators are represented as full QM atoms, there is a possibility that the bulk of the excited state might be on a spectator molecule (for example if the active molecule is an electron donor and the spectator is an electron acceptor). UV/Vis spectra are generated for each molecule and associated with its output entry. The (unweighted) average spectrum is also generated and associated with the output entry that contains the entire system. As the molecules are thermally equilibrated, the unweighted spectrum should include the effect of the Boltzmann distribution.
The structures for each cluster are incorporated into the project as an entry group, with the original cell containing all the molecules as the first entry in the group. The cluster entries are named with the same convention as the output file. By fixing the first entry in the Workspace, then including different clusters, you can identify the location of each active molecule in the amorphous structure, and correlate the environment of the active molecule with its properties.
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
Amorphous Properties Panel Features
- Use structures from option menu
- Open Project Table button
- File name text box and Browse button
- Limit active molecules to those of type option and menu
- Nearest neighbor distance option and text box
- Isolated molecules option
- Options button
- Treat spectator atoms as option menu
- Molecule subset options
- Property options
- Bonds to optimize option menu
- Other keywords text box
- Job toolbar
- Status bar
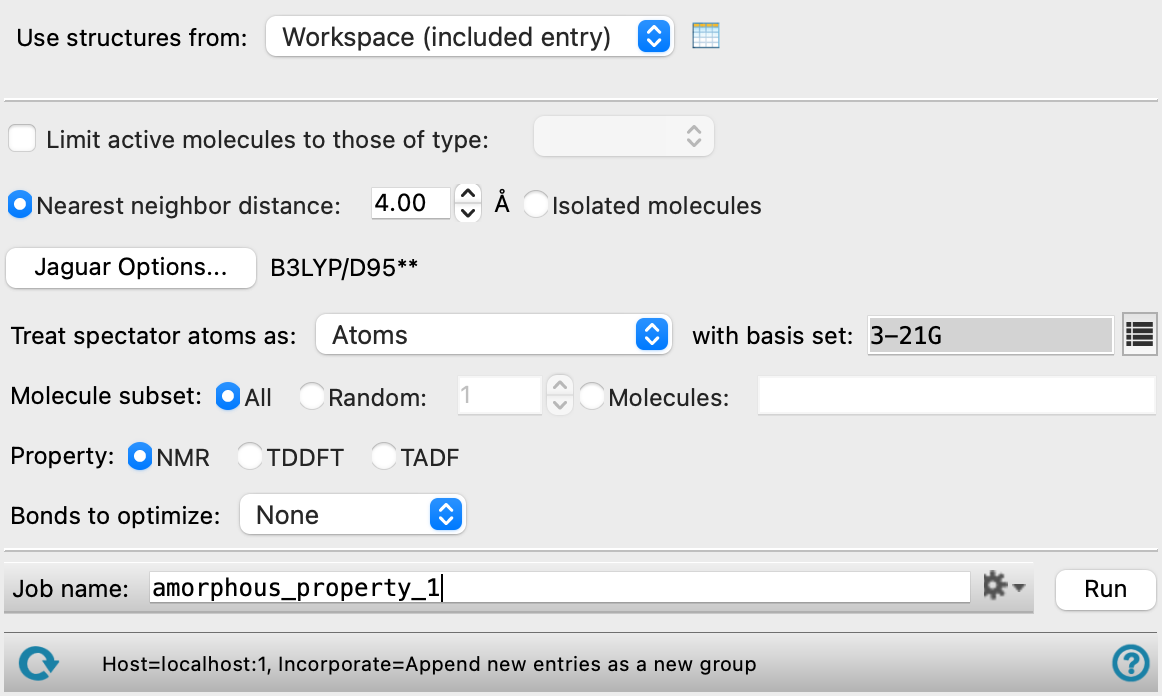
- Use structures from option menu
-
Choose the structure source for the current task.
- Workspace (included entry)—Use the entry that is currently included in the Workspace. Only one entry must be included in the Workspace.
- File—Use the specified file. When this option is selected, the File name text box and Browse button are displayed.
- Open Project Table button
-
Open the Project Table panel, so you can

- File name text box and Browse button
-
Enter the file name in this text box, or click Browse and navigate to the file. The name of the file you selected is displayed in the text box.
- Limit active molecules to those of type option and menu
-
Limit the active molecules to those of the type selected from the option menu. The input is analyzed to find each unique molecule type, and these are listed on the option menu by molecular formula and number of molecules.
- Nearest neighbor distance option and text box
-
Select this option to specify the distance from the active molecule to any neighbor molecule to be included as a spectator. A neighbor is included if any atom in the neighbor is within the specified distance of any atom in the active molecule.
- Isolated molecules option
-
Select this option to run the calculations on isolated molecules, without any neighbors included.
- Options button
-
Set Jaguar options for the amorphous properties calculations. Opens the Jaguar Options - Amorphous Properties Calculations Dialog Box. This dialog box also allows you to specify additional Jaguar keywords. The solvent (if any), level of theory, and basis set are shown to the right of the button.
- Treat spectator atoms as option menu
-
Choose how to treat spectator atoms in the Jaguar calculations for the neighbors. There are three options:
-
Atoms—Include the full atoms with all their electrons in the Jaguar calculations (quantum mechanical treatment). You can specify a different basis set from the active molecule in the Basis set text box (see below).
-
Existing partial charges—Replace the atoms with the partial charge taken from a Maestro property. The property can be chosen from the Atom property option menu (see below).
-
ESP charges—Replace the atoms with partial charges derived from the electrostatic potential in a single-point B3LYP Jaguar calculation on a sample spectator molecule. You can specify the basis set for the ESP charge calculation in the Basis set text box (see below).
If either of the options for charges is chosen, the spectator molecules are replaced with point charges in the Jaguar calculation on the active molecule. This makes the calculation faster, but may lose some of the value of having distributed electronic charge in the actual spectator molecules.
- Basis set text box
-
Jaguar keyword for the basis set used for the spectator molecules, either when treated as atoms or as point charges using ESP charges from Jaguar. When you click in the text box, a small panel is displayed in which you can select the basis set and add diffuse and polarization functions. You can also edit the keyword directly. See Basis Sets for a list of basis set keywords.
- Atom property option menu
-
Choose the atom property that defines the partial charges from the option menu.
-
- Molecule subset options
-
Choose an option for the subset of molecules whose properties are to be calculated. These options are affected by the Limit active molecules to those of type option.
-
All—Do calculations on all molecules in the cell, or on all molecules of the chosen type.
-
Random—Choose a subset of molecules at random. The number of molecules to choose can be specified in the text box. The molecules are chosen from those of the specified type, or from all molecules if no type is specified.
-
Molecules—Specify the molecules whose properties you want to calculate, by providing a comma-separated list of molecule numbers in the text box. If you specify a particular type of molecule for the active molecule, you must ensure that the list contains only molecules of that type.
-
- Property options
-
Choose a property to calculate.
-
NMR—Calculate NMR shieldings.
-
TDDFT—Calculate singlet excitation energies and oscillator strengths by TDDFT.
-
TADF—Calculate singlet and triplet excitation energies, singlet oscillator strengths, and the lowest S1-T1 gap by TDDFT.
-
- Bonds to optimize option menu
-
Choose the bonds whose lengths are to be optimized before calculating the properties. You can optimize all bonds (All), just bonds to hydrogens (To hydrogens), or bypass optimization (None). This option is only available for NMR calculations, as these are particularly sensitive to geometry.
- Other keywords text box
-
Specify other Jaguar keywords for the property calculation, as keyword=value pairs separated by spaces. These are intended for control of things like the accuracy and grid: you cannot use the dftname, basis, igeopt, or nmr keywords here. The keywords do not apply to the calculation of ESP charges for spectator atoms. See The gen Section of the Jaguar Input File for information on Jaguar keywords.
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Amorphous Properties Calculations - Job Settings Dialog Box, where you can make settings for running the job.
- Status bar
-
The status bar displays information about the current job settings and status for the panel. The settings includes the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
Use the Reset button
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel. The status bar also contains the Help button
 , which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a
, which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a  button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.
button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.