Bond and Ligand Dissociation Panel
Calculate the change in energy for dissociating molecules into combinations of two fragments that come from breaking covalent non-ring single bonds or removing ligands from a metal atom. All possible fragments or a restricted set of fragments can be selected for calculation.
The dissociation is heterolytic if the neutral fragments are closed-shell molecules, otherwise it is homolytic. Thus, for carbon-carbon bonds and bonds to hydrogen, the dissociation is homolytic and produces two radicals, whereas for CO bonding to metals, the dissociation is heterolytic.
To open this panel, do one of the following:
- Click the Tasks button and browse to Quantum Mechanics → Bond and Ligand Dissociation
- Click the Tasks button and browse to Materials → Quantum Mechanics → Workflows → Bond and Ligand Dissociation
The following licenses are required to use this panel: MS Maestro, Jaguar, MS Force Field Applications (optional)
- Using
- Features
- Additional Resources
Using the Bond and Ligand Dissociation Panel
When specifying the bonds to break, you can apply restrictions on top of the choice of a single acyclic bond. You can include bonds to hydrogen along with other atoms, you can restrict the choice to break only bonds to hydrogen, or you can exclude bonds to hydrogen. As well as restricting bonds to hydrogen, there is a more general feature for either including or excluding bonds based on whether any atom in the bond is in a match to a set of SMARTS patterns.
The calculations are run with Jaguar, and the geometry of each reactant and product is optimized. To maximize efficiency, a unique set of fragments for the entire input is assembled, and calculations are performed on the unique fragments. However, if the products are frozen at the reactant geometry to produce vertical bond dissociation energies, all fragments are calculated, as there is no guarantee that fragments with the same connectivity have the same geometry.
This means, for example, that if a phenyl ring occurs in more than one place either within a single molecule or in different molecules, only one calculation is performed on the phenyl ring. Likewise, dissociation from a singlet and a triplet state produce the same set of fragments, so the cost of calculating both is not much more than that of calculating one of the initial states. (The extra cost is just the cost of the extra initial states).
You can calculate the bond dissociation energy for single, acyclic bonds, and polydentate ligands in the same run. In the case of ligands, the bond dissociation energy is the sum of the energies required to break all metal-ligand bonds.
The results of the bond dissociation calculation are returned as three sets of entries:
-
Reaction entries, in the main project group for the job. Each entry contains the reactant and product structures for a reaction. They are sorted by reactant, then by increasing bond dissociation energy.
The reaction energy is added as an entry property, the broken bond is marked on the reactant structure, and the atoms on each end of the broken bond are marked on the product structures. The bond dissociation energy for the breaking bond can be displayed as a bond label (Workspace → Bond Labels).
The reactant structure is placed with the broken bond on the x axis, on the negative end. The product structures are placed on the positive side of the x axis, and displaced in the y direction so they don't overlap. They have the same orientation as in the reactant structure.
For polydentate ligands, each atom that ligates the metal is marked, and each metal-ligand bond is marked in the reactant; the average of the vectors for the broken bonds is aligned on the x axis in this case.
-
Reactant structures, one per entry, in a subgroup labeled Reactants. These are the optimized input structures. (If optimization is turned off, these are the unaltered input structures.)
When included in the Workspace, these structures show labels on each dissociating bond that give the bond dissociation energy for the bond or ligand. In the case of ligands, the energy is the sum of all metal-bond breaking energies for that ligand. The labels can be turned on or off in the Bond Labels Panel (Workspace → Bond Labels), or by clicking the Workflow Action button
 for the entry; or they can be shown and hidden by using the Annotations button on the Workspace Configuration toolbar.
for the entry; or they can be shown and hidden by using the Annotations button on the Workspace Configuration toolbar.
In addition, the weakest bond energy is added as an entry property to each reactant structure.
-
Fragment structures, one per entry, in a subgroup labeled Fragments. These are the optimized fragment structures. They are titled "fragment # X", where # is the index of the fragment and X gives information about the state. The job name is also appended to the title, so that you can distinguish the results of different runs.
The job directory also contains .svg files for each unique reactant and starting state (s0, t1, s1, anion, cation). Each .svg file shows a 2D rendering of the molecule with the weakest bond highlighted. SVG files can be viewed by most image viewers.
If a job fails due to problems like network or hardware issues, you can restart the job with the Restart Workflow panel.
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
Bond and Ligand Dissociation Panel Features
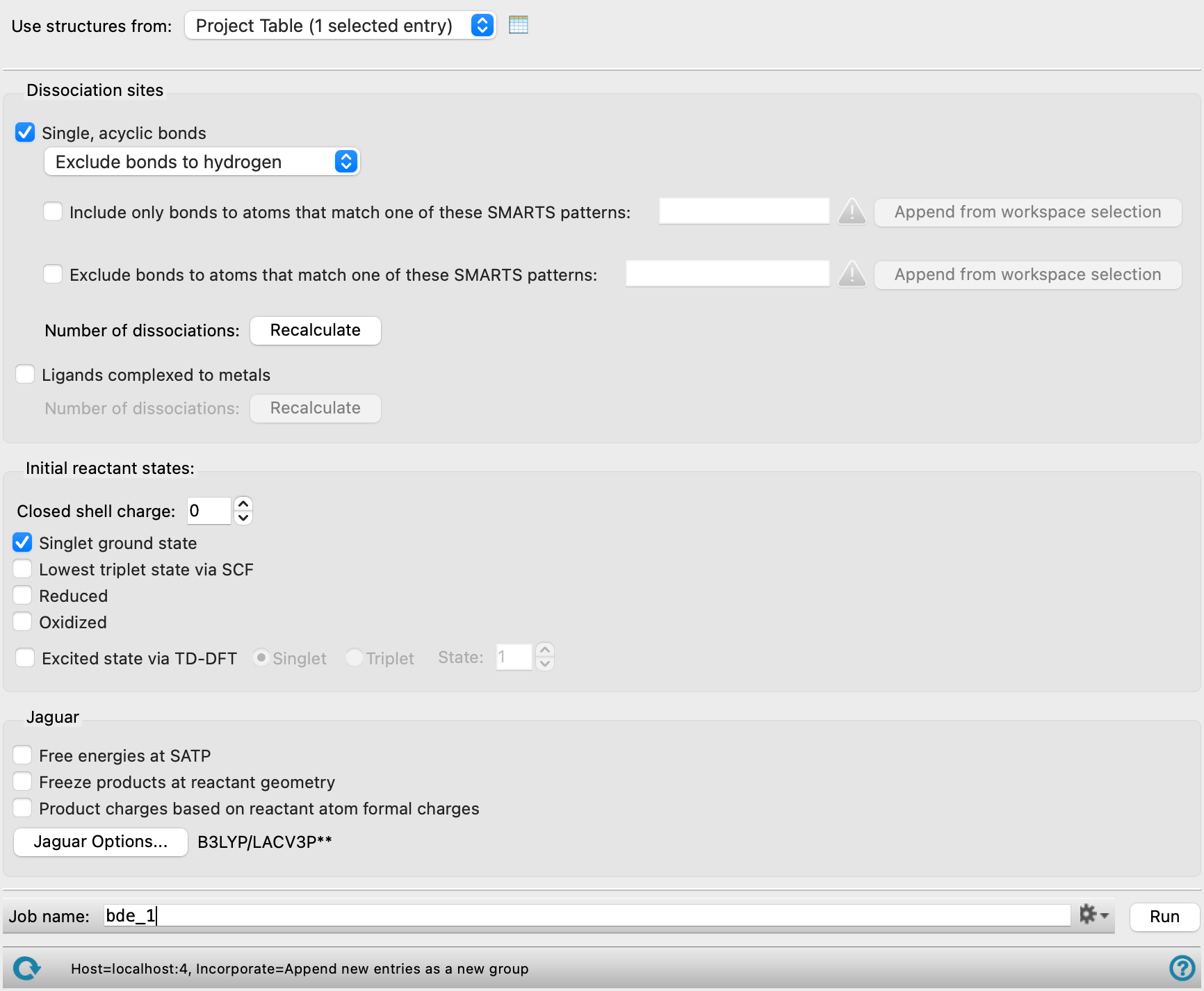
- Use structures from option menu
-
Choose the structure source for the current task.
- Project Table (selected entries)—Use the entries that are currently selected in the Project Table.
- Workspace (included entries)—Use the entries that are currently included in the Workspace, treated as separate structures.
- File—Use the specified file. When this option is selected, the File name text box and Browse button are displayed.
- File name text box and Browse button
-
Enter the file name in this text box, or click Browse and navigate to the file. The name of the file you selected is displayed in the text box.
- Dissociation sites section
-
Specify the kinds of dissociation reaction to be considered and the sites to consider for the reaction. At least one of the options (Single, acyclic bonds or Ligands complexed to metals) must be selected.
- Single, acyclic bonds option
-
Calculate dissociation energies for the homolytic breaking of all single bonds in each molecule that are not part of a ring.
By default, bonds to hydrogen are not included. If you want to consider bonds to hydrogen as well, choose Include bonds to hydrogen from the option menu. If you want to consider only the bonds to hydrogen, i.e. to calculate hydrogen abstraction energies, choose Include only bonds to hydrogen from the option menu.
As well as controlling bonds to hydrogen, you can control bonds to other atoms or groups that match a SMARTS pattern:
-
Include only bonds to atoms that match one of these SMARTS patterns—Bonds will only be considered for breaking if one of the atoms in the bonds is in a match to the SMARTS patterns listed in the text box. You can include multiple SMARTS patterns, separated by spaces.
-
Exclude bonds to atoms that match one of these SMARTS patterns—Bonds will not be considered for breaking if either of the atoms in the bonds is in a match to the SMARTS patterns listed in the text box. You can include multiple SMARTS patterns, separated by spaces.
Click Recalculate to enumerate the bonds. This can take some time if there are a lot of bonds. Text showing the number of bonds found replaces this button when the enumeration is done. If you change the settings in this section, the button is redisplayed and must be clicked again to enumerate the bonds.
If the molecule is a metal complex, bonds between monodentate ligands and the metal are included as well as bonds in the ligands.
-
- Ligands complexed to metals option
-
Calculate dissociation energies for the removal of single ligand molecules that are complexed to metals. This may involve the breaking of more than one metal-ligand bond, if the ligand is polydentate. The dissociation is heterolytic if the fragments are closed-shell species: this allows for the dative bonding that is typical of many ligands; however any dissociation reaction that produces H+ is ignored.
Click Recalculate to enumerate the ligands. This can take some time if there are a lot of ligands. Text showing the number of ligands found replaces this button when the enumeration is done. If you change the settings in this section, the button is redisplayed and must be clicked again to enumerate the ligands.
- Initial reactant states section
-
Select options for the initial state of the molecules you want to dissociate. You can select more than one state, which produces multiple results for each molecule. The input molecule must be a closed-shell singlet. This includes transition-metal complexes. The number of reactions depends on the charge of the initial state: if the charge is not zero, two reactions are generated, with the charge on one fragment or the other; if the charge is zero only one reaction is generated, with neutral fragments.
- Closed shell charge box
-
Specify the charge on the closed-shell input molecule. The allowed values are 1, 0, and −1 unless the Product charges based on reactant atom formal charges option is selected.
- Singlet ground state option
-
Calculate dissociation energies from the ground state of the input molecule.
- Lowest triplet state by SCF option
-
Calculate the dissociation energies from the lowest triplet state of the input molecule, as determined from an SCF calculation.
- Reduced option
-
Calculate dissociation energies from the reduced doublet state, i.e. with one more electron. This option is not allowed if the charge on the input molecule is −1 and the Product charges based on reactant atom formal charges is not selected.
- Oxidized option
-
Calculate dissociation energies from the oxidized doublet state, i.e. with one less electron. This option is not allowed if the charge on the input molecule is +1 and the Product charges based on reactant atom formal charges is not selected.
- Excited state via TD-DFT options and text box
-
Calculate the dissociation energies from the chosen excited state of the input molecule. Select the spin for the state, from Singlet or Triplet, and enter the excited state number in the State text box.
- Jaguar options section
-
Specify options for the Jaguar calculations.
- Free energies at SATP option
-
Calculate free energies of dissocation at standard ambient temperature and pressure (298.15 K and 1 atm). As this involves frequency calculations for each structure, the time taken is substantially longer.
If you want to calculate the free energies for specific isotopes, you will need to set the isotopic mass numbers as atom properties on the structures. See Changing Atom and Residue Properties for information on setting isotopic mass numbers.
- Freeze products at reactant geometry option
-
Run the calculations on the products at the reactant geometries, to obtain vertical bond dissociation energies. As fragments of the same connectivity can have different geometries and so can't necessarily be reused, calculations are run on all fragments, even if redundant.
- Product charges based on reactant atom formal charges option
-
Select this option to enforce that the charge on each product fragment is the sum of the formal charges on the atoms in the fragment. The formal charges must be set prior to running the calculation. Selecting this option removes the limitations on the Closed shell charge. For the reactant structure, formal charges must sum up to the charge on the reactant set in the Initial reactant states.
- Options button
-
Set Jaguar options for the dissociation energy calculations. Machine learning force fields can be used in place of the quantum mechanics calculations. Opens the Jaguar Options - Bond and Ligand Dissociation Dialog Box. This dialog box also allows you to specify additional Jaguar keywords. The solvent (if any), level of theory, and basis set are shown to the right of the button.
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Bond and Ligand Dissociation - Job Settings Dialog Box, where you can make settings for running the job.
- Status bar
-
The status bar displays information about the current job settings and status for the panel. The settings includes the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
Use the Reset button
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel. The status bar also contains the Help button
 , which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a
, which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a  button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.
button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.