MxMD Simulation Details
System Setup
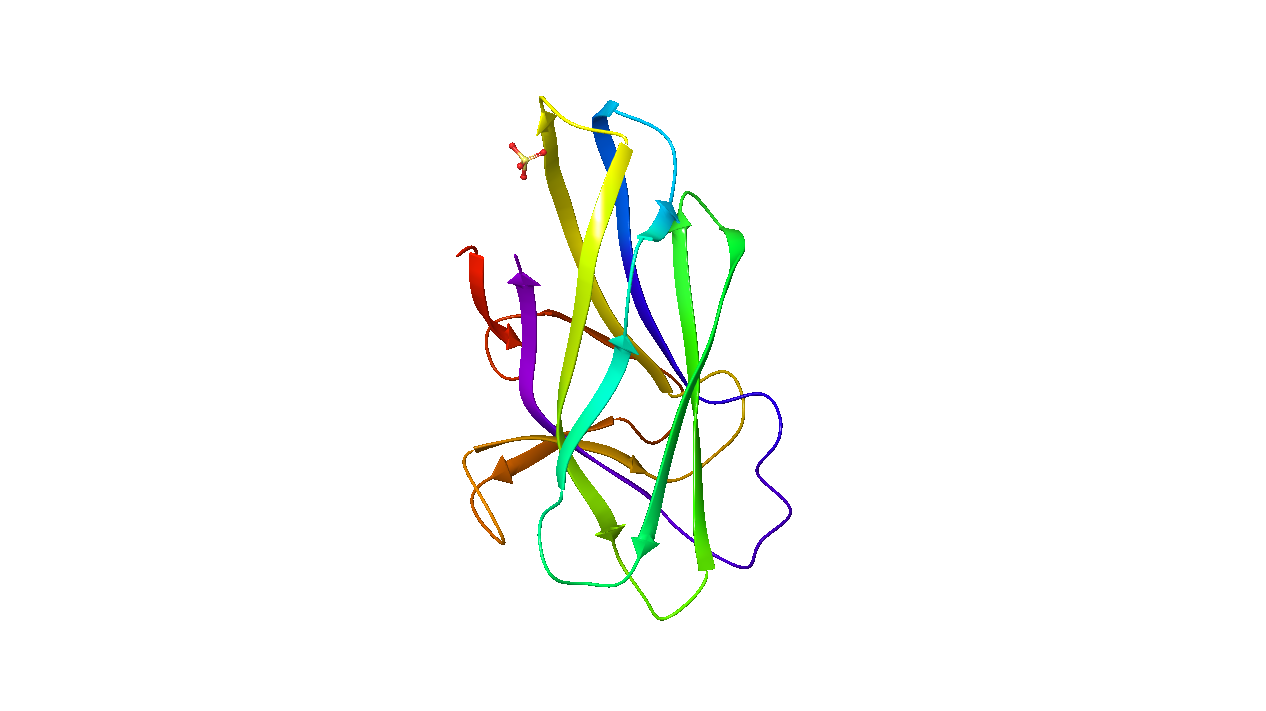
By default, ten cosolvent/water systems are generated for each probe. Each system contains the input structure of the protein or DNA/RNA target (solute), a 7 Å layer of organic cosolvent and water molecules. The number of cosolvent molecules is tuned to be approximately 5% volume by volume.
|
|
+ |
|
+ |
|
= |
|
|
(Protein, DNA, or RNA) |
7Å cosolvent layer |
Water |
Cosolvent systems |


The default procedure to generate the cosolvent/water mixture is as follows:
-
Solvate the input solute structure with the cosolvent system.
-
Remove all cosolvent molecules that overlap with the solute, or that are farther than 7 Å away from the solute.
The overlap is determined by scaling the van der Waals radius of the probe by the factor svdw:

where
 is the number of heavy atoms in the probe. Thus, larger probes have a greater scaling factor.
is the number of heavy atoms in the probe. Thus, larger probes have a greater scaling factor. -
Solvate the solute/cosolvent system in water with a solvent buffer of 15 Å.
-
Shrink the size of the water box to match the target ratio of cosolvent to water (5% volume by volume).

where Nwat and Nprobes are the number of water and probe molecules; mwat and mprobe are the masses of one water and probe molecule; vvtarget is the target volume by volume (v/v) ratio; and ρprobe is the density of the probe’s homogeneous mixture at 300 K.
System Setup for Membrane Proteins
The procedure to generate the cosolvent/water mixture for a membrane protein is as follows:
-
Solvate the input protein structure with the cosolvent system.
-
Place the membrane bilayer and waters around the protein.
-
Remove all cosolvent molecules that overlap with the protein.
The overlap is determined by scaling the van der Waals radius of the probe by the factor svdw:

where
 is the number of heavy atoms in the probe. Thus, larger probes have a greater scaling factor.
is the number of heavy atoms in the probe. Thus, larger probes have a greater scaling factor.Cosolvent molecules that are farther than 7 Å away from the membrane or protein are also removed.
-
Remove all the cosolvent molecules that overlap with the lipids.
-
Remove all the waters that overlap with the cosolvent molecules.
-
Shrink the size of the water/lipid box to match the target ratio of cosolvent to water (5% volume by volume).

where Nwat and Nprobes are the number of water and probe molecules; mwat and mprobe are the masses of one water and probe molecule; vvtarget is the target volume by volume (v/v) ratio; and ρprobe is the density of the probe’s homogeneous mixture at 300 K.
MxMD Simulation Protocol
The cumulative simulation time for each cosolvent/water system is approximately 20 ns by default. The first 15 ns are used for equilibrating the system. The last 5 ns are used for data collection. The simulation protocol consists of the following stages:
- 24 ps Brownian Dynamics, NVT, T = 10 K, timestep = 1 fs, restraints on all solute atoms
- 24 ps Brownian Dynamics (MD), NVT, T = 10 K, timestep = 1 fs, restraints on solute heavy atoms
- 12 ps MD, NVT, T = 10 K, timestep = 1 fs, restraints on solute heavy atoms
- 12 ps MD, NPT, T = 10 K, p = 1.013 bar, timestep = 2 fs, restraints on solute heavy atoms
- 12 ps MD, NPT, T = 300 K, p = 1.013 bar, timestep = 2 fs, restrains on solute heavy atoms
- 15 ns MD, NPT, T = 300 K, p = 1.013 bar, timestep = 2 fs, no restraints
- 5 ns MD, NPT, T = 300 K, p = 1.013 bar, timestep = 2 fs, no restraints
The last stage is used to collect data. A trajectory frame is written every 4.8 ps, corresponding to approximately 1000 trajectory frames per cosolvent/water simulation for the default simulation time. The trajectory is written in XTC format. See Trajectory Player for more information.