Protein Structure Alignment Panel
Bring two or more proteins into a common frame of reference by structurally aligning (superimposing) them, taking into account secondary structure.
To open the Protein Structure Alignment panel: click the Tasks button and browse to Structure Alignment → Protein Structure Alignment.
- Using
- Features
- Additional Resources
Using the Protein Structure Alignment Panel
It is often difficult to superimpose protein structures. Reasons include:
- low structural similarity
- low (or no) sequence identity
- a different number of residues
- different sequence numbering
This panel greatly facilitates the superimposition of protein structures with an algorithm that attempts to align secondary structure elements.
Structure alignment operates on project entries that have been included in the Workspace or are selected in the Project Table. You can choose which to use from the Use proteins from option menu. If you choose Workspace (included entries) the reference structure, whose frame of reference is used for the alignment, must be the first included entry (the entry with the lowest row number). To make another protein the reference, move that protein above all the other included entries in the Project Table. If you choose Project Table (selected entries), the reference structure must be the only structure included in the Workspace.
By default, the alignment includes all residues. However, it is often useful to align a subset of residues that have greater similarity than the structures as a whole, or are more informative to compare. Because this program uses matching of secondary structure elements, the subset should have enough contiguous residues to include at least one helix or strand. Very small numbers of residues do not produce useful alignments.
You might need to try different subsets to obtain the most useful alignment. You can use the picking controls, choose a predefined set of atoms from the selection button menu, or open the Atom Selection Dialog Box (ASD) to specify subsets. In this dialog box you can select a range of residue numbers, specify portions of the residue sequence, or click Secondary Structure in the list of options on the left to choose residues based on their location in Helix, Strand, or Loop structures. It might also be helpful to display the sequence viewer with the secondary structure assignment, and pick residues in the sequence viewer.
When the alignment calculation is complete, the structures are placed in the same frame of reference in the Workspace, and the results of the calculation are displayed in the Protein Structure Alignment Results panel. The aligned residues are listed and an RMSD and Alignment Score are reported. The RMSD is calculated based only on those residues that are considered to have been successfully aligned, and therefore does not represent an overall comparison of the structures. It is computed from the C-alpha atoms of the aligned residues.
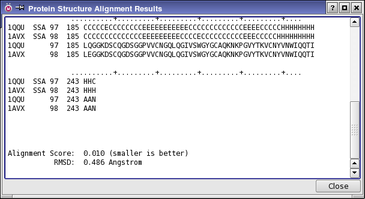
An Alignment Score lower than 0.6–0.7 indicates a good alignment. Alignment Scores greater than about 0.7–0.8, or a failure of the structural alignment calculation, indicates there is insufficient structural similarity for a meaningful alignment. For details on the algorithm, see the paper by Honig and Yang (J. Mol. Biol.2000, 301, 665).
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
Protein Structure Alignment Panel Features
- Use proteins from option menu
- Reference residues picking controls
- Residues to align options and picking controls
- Align button
- Force alignment option
- Use proteins from option menu
-
Choose the source of the proteins to align, from Workspace (included entries) or Project Table (selected entries). The proteins are aligned to the frame of the project entry that has the smallest row number.
- Reference residues picking controls
-
Specify the residues in the reference protein to which the other proteins will be aligned, using the standard picking tools. You should specify residues that contain at least one secondary structure element. You must pick residues in the reference protein, which is the project entry with the smallest row number (highest in the table). A warning is displayed if you pick residues in another protein.
- Residues to align options and picking controls
-
Specify the residues in the other proteins that are to be aligned to the reference residue set.
-
Use same ASL as reference residues—If you restricted the residues in the reference protein, select this option to use the same expression for the proteins to be aligned as for the reference. This does not necessarily mean the exact same residues (although it can), because the ASL expression is evaluated independently for each protein being aligned.
-
Use separate ASL—Select this option to specify the subset with a different residue selection (ASL expression) from the reference, and use the picking tools to make the selection. You might need to try different subsets to obtain the most useful alignment. Subsets should include at least one secondary structure element (helix or strand). Clicking the Common objects button then choosing Select opens the Atom Selection dialog box to the Residue tab. Here you can select a range of residue numbers, specify portions of the residue sequence, or click Secondary Structure in the list of options on the left to choose residues based on their location in Helix, Strand, or Loop structures. Subsets can be combined, intersected, or otherwise modified in the Atom Selection dialog box.
-
- Align button
-
Click this button to start the Protein Structure Alignment job. The Protein Structure Alignment Results panel opens, showing the text "Calculating alignment..." until the alignment is done, when the text is replaced by the results. The text in the results panel is replaced each time you click Align.
- Force alignment option
-
Align proteins even if the structures are not sufficiently similar.