Create Reaction Network Profiler Input Structures Panel
Prepare input for the structures at each stage of a reaction network, annotated with the relevant properties. The prepared structures can then be used in the Reaction Network Profiler Panel.
To open this panel: click the Tasks button and browse to Materials → Quantum Mechanics → Reaction Network → Create Reaction Network Profiler Input.
The following licenses are required to use this panel: MS Maestro
- Using
- Features
- Additional Resources
Using the Create Reaction Network Profiler Input Structures Panel
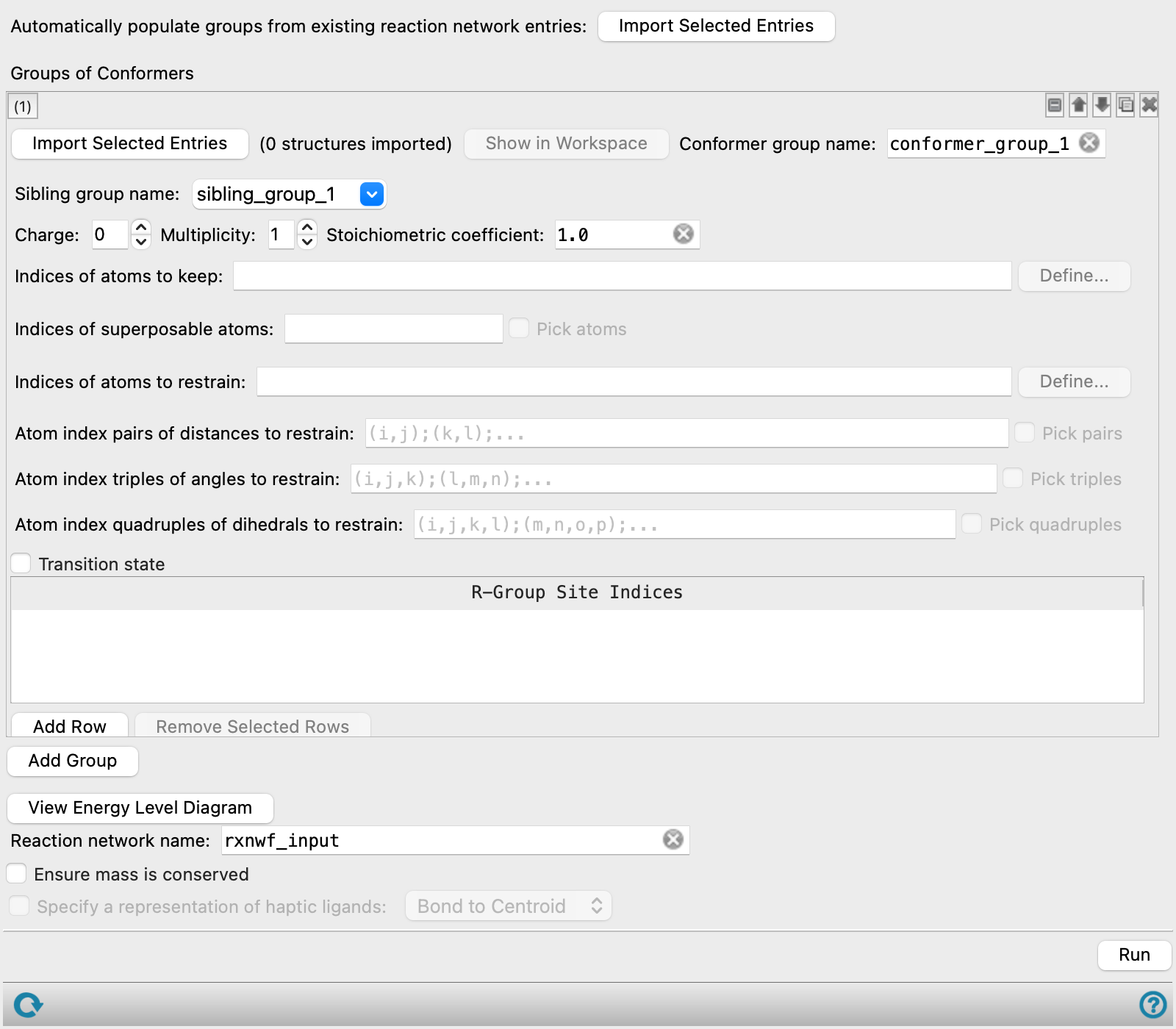
This panel sets up structures for the reaction network, in which the structures are optimized to locate a minimum or a transition state. The main purpose of the panel is to specify restraints on atoms or geometric parameters in the process of finding the minimum or the transition state. These restraints (and other settings) are stored as entry properties in the output structures.
The panel operates on groups of conformers. Each group must have the same connectivity and atom numbering, but you can include stereoisomers (R,S or E,Z) as well as different geometries of rotatable groups. The group can of course consist of a single structure. Settings are made for each group of conformers, and the groups are stored in the Project Table.
The controls for these settings can be added for a new group (click Add Group), or duplicated from an existing group (click the Duplicate button  ). The order of the groups is irrelevant to the calculations done in the reaction network: each group is processed independently. You can therefore rearrange the groups as you like, using the tools at the top right of the group controls.
). The order of the groups is irrelevant to the calculations done in the reaction network: each group is processed independently. You can therefore rearrange the groups as you like, using the tools at the top right of the group controls.
A stage in the reaction network can consist of multiple conformer groups. The conformer groups in a stage are "siblings": they have the same parent stages (and also the same child stages). This allows you to carry a structure through several reaction stages where it is a spectator until it becomes a reactant in a particular stage, and likewise to carry a product or byproduct through subsequent stages of the reaction. Introducing the spectators allows you to define stages that have the same stoichiometry, which in turn allows you to compare energies at each stage to obtain a reaction profile.
For transition states, a preoptimization is performed with Jaguar prior to doing the transition state search, using specified distance, angle, and dihedral restraints. In the transition state search, these restrained coordinates become active coordinates that are used to guide the transition state search, which is performed without restraints. In this way, the non-active coordinates can be optimized before the transition state is performed.
For reactants, products, and intermediates, where the optimization is to a minimum, it is usually not a good idea to apply restraints, as only a single optimization step is performed.
Create Reaction Network Profiler Input Structures Panel Features
- Import Selected Entries button
- Groups of Conformers section
- Conformer group tools
- Import from Selected Entries button
- Show in Workspace button
- Conformer group name text box
- Sibling group name text box
- Add parent group name option menu, Add button, text box, and Clear button
- Charge text box
- Multiplicity text box
- Stoichiometric coefficient text box
- Indices of atoms to keep text box and Define button
- Indices of superposable atoms text box and Pick atoms option
- Indices of atoms to restrain text box and Define button
- Atom index pairs of distances to restrain text box and Pick pairs option
- Atom index triples of angles to restrain text box and Pick triples option
- Atom index quadruples of dihedrals to restrain text box and Pick quadruples option
- Transition state option
- R-Group Site Indices table
- Add Row button
- Remove Selected Rows button
- Add Group button
- View Energy Level Diagram button
- Reaction network name text box
- Ensure mass is conserved option
- Specify a representation of haptic ligands option and menu
- Run button
- Status bar
- Import Selected Entries button
-
Import an entire network from the Project Table. All the fields in the panel are populated. You must select the entry group containing the network first. Importing a network allows you to edit it to replace the existing network or create a new network (by changing the reaction network name).
- Groups of Conformers section
-
In this section, you can set up the input for structures at each stage of a reaction network. Each stage (reactant, transition state, intermediate, product) could have multiple structures - for example, you may have two reactants. For each structure there could be different conformations that potentially contribute to the reaction sequence. This section provides a set of tools that can be duplicated to allow you to specify input settings for the conformers of each structure. New entries in a new entry group are created for the conformers, with the settings are stored as properties. Note that you can specify a single structure—it is not necessary to have conformers.
- Conformer group tools
-
This set of tools allows you to make settings for the conformers of a particular structure in reaction network, here referred to as a "conformer group". The settings enable you to specify atoms to keep and superimpose in subsequent uses of the Swap Fragments Panel, atoms to restrain in the MacroModel conformational search on the structure that can be done prior to optimization, and distances, angles, and dihedrals to restrain for the Jaguar (QM) preoptimization of transition states.
- conformer group management buttons
-
These buttons perform display and ordering operations on the conformer group. They allow for easy duplication and rearrangement of conformer groups.


Show or hide the contents of the conformer group. When hidden, only the conformer group number, label (if any) and these buttons are displayed. This is useful when you have a number of conformer groups and want to compare two separate conformer groups, for example. 
Delete the conformer group. The order of the groups does not matter, so you can move groups next to each other to make it easier to copy data from one to another.
- Import from Selected Entries button
-
Import the conformer group from the entries that are selected in the Project Table. The selected entries must be conformers, i.e. have the same connectivity and atom numbering. You can have stereoisomers (R,S or E,Z) among the conformers as well as different geometric arrangements of rotatable groups. The entries must also have the multiplicity and molecular charge set as properties (see Add Standard Molecular Property Panel for more information).
- Show in Workspace button
-
Display the first conformer in the Workspace, so that you can perform picking for distances, angles, or dihedrals to restrain.
- Conformer group name text box
-
Enter the name you want to give to the new entry group that contains the conformer structures. The group is created when you click Run, and is added to the parent group, as specified below.
- Sibling group name text box
-
Specify the sibling group (reaction stage) to which the current conformer group belongs. You can enter the name in the text box, or click the arrow to choose from already defined sibling group names.
- Add parent group name option menu, Add button, text box, and Clear button
-
Specify the parent stages (one or more) for this stage, by sibling group name. Choose a parent from the option menu and click the Add button to add the parent. The option menu is populated from the Sibling group name fields of the conformer groups. The group name is displayed in the text box. To remove all parent names, click Clear.
Any stage can have multiple parents, and multiple stages can have the same parent. The parent information is used in the reaction network to identify the flow, which is useful when the reaction network scheme is not linear, e.g. there are branches in the reaction network scheme.
- Charge text box
-
Specify the total charge of each structure in the conformer group, for the Jaguar calculations. If the conformer structures consist of multiple molecules, only the total charge is set, not the charge of individual molecules, as the structure is treated as a single structure by Jaguar. The charge can change between parent and child conformer groups, as the stages are not required to be part of a single reaction sequence in which conservation holds.
- Multiplicity text box
-
Specify the total spin multiplicity of each structure in the conformer group, for the Jaguar calculations. If the conformer structures consist of multiple molecules, only the total multiplicity is set, not the multiplicity of individual molecules, as the structure is treated as a single structure by Jaguar. The multiplicity can change between parent and child conformer groups, as the stages are not required to be part of a single reaction sequence in which conservation holds.
- Stoichiometric coefficient text box
-
Specify the stoichiometric coefficient by which to multiply the mass and energy of the given structure in order to get effective values of these properties. This can be useful for when getting an accurate energy of a desired structure requires a symmetrical arrangement of multiple such structures. The stoichiometric coefficients are used when the Ensure mass is conserved option is selected. The stoichiometric coefficients must give no fractional atoms to be valid.
- Indices of atoms to keep text box and Define button
-
Specify the atoms to keep in this structure when swapping fragments in the Swap Fragments Panel. These atoms do not need to be specified to run the reaction network (which does not use these data), but they are useful if you are planning to prepare another set of structures based on this structure for the reaction network.
You can enter the atom indices as a comma-separated list, or click Define to use the standard Picking Tools to select the atoms.
- Indices of superposable atoms text box and Pick atoms option
-
Specify the atoms in this structure to superimpose on another structure when swapping fragments in the Swap Fragments Panel. These atoms do not need to be specified to run the reaction network (which does not use these data), but they are useful if you are planning to prepare another set of structures based on this structure for the reaction network.
You can enter the atom indices as a comma-separated list, or select Pick atoms to pick the atoms in the Workspace. The order of the atom indices is important: the atom pairs to be superimposed are taken by matching the lists of superimposable atoms in the order given.
- Indices of atoms to restrain text box and Define button
-
Specify the atoms to restrain in the MacroModel conformational search that can be performed on this structure in the Reaction Network Profiler Panel. In addition to restraining parts of the structure that you don't want to vary in the conformational search, you should specify any atom that does not have (good) force field parameters or are not treated well by the force field, such as metal atoms and eta-bonded ligand atoms.
You can enter the atom indices as a comma-separated list, or click Define to use the standard Picking Tools to select the atoms.
- Atom index pairs of distances to restrain text box and Pick pairs option
-
Specify distances to restrain in the quantum-mechanical optimization of the structure. Enter the atom pairs for the distances in the text box in the format (i,j);(k,l);... or select Pick pairs and pick pairs of atoms in the Workspace to define the distances to restrain. For a transition state, these distances are frozen in the preoptimization of the structure, and then converted to active coordinates for the transition state search. For other structures, it is not recommended to apply restraints, as only a single optimization is performed.
- Atom index triples of angles to restrain text box and Pick triples option
-
Specify angles to restrain in the quantum-mechanical optimization of the structure. Enter the atom triples for the angles in the text box in the format (i,j,k);(l,m,n);... or select Pick triples and pick three atoms in the Workspace for each angle you want to restrain. For a transition state search, these angles are frozen in the preoptimization of the structure, and then converted to active coordinates for the transition state search. For other structures, it is not recommended to apply restraints, as only a single optimization is performed.
- Atom index quadruples of dihedrals to restrain text box and Pick quadruples option
-
Specify dihedrals to restrain in the quantum-mechanical optimization of the structure. Enter the atom index quadruples for the dihedrals in the text box in the format (i,j,k,l);(m,n,o,p)... or select Pick quadruples and pick four atoms in the Workspace for each dihedral you want to restrain. For a transition state search, these dihedrals are frozen in the preoptimization of the structure, and then converted to active coordinates for the transition state search. For other structures, it is not recommended to apply restraints, as only a single optimization is performed.
- Transition state option
-
Mark this structure as a transition state structure. In the reaction network, a transition state search is performed if this option is set, otherwise a minimization is performed.
- R-Group Site Indices table
-
Specify R-group enumeration sites as a pair of bonded atoms. Use the Add Row button to add a row. In each row, enter the (from, to) atom index pairs in the text box in the format (i,j);(k,l);... or select Pick pairs and pick pairs of atoms in the Workspace to define the enumerate site. Pairs to be used with same set of enumeration groups should be listed in the same row. Pairs to be used with different sets of enumeration groups should be listed in different rows.
- Add Row button
-
Add a row to the R-Group Site Indices table.
- Remove Selected Rows button
-
Remove selected rows in the R-Group Site Indices table. Select one or more rows by clicking on the row number.
- Add Group button
-
Add another set of conformer group controls, below the last one.
- View Energy Level Diagram button
-
Click to view a mockup energy level diagram with fictitious relative energies. This is helpful in checking for a proper reaction network prior to running a job. Click Save to open a file selector, to name and save the file. Click Close to exit the plot.
- Reaction network name text box
-
Name of the reaction network to which all the conformer groups are added. This defines a "super group". You can use the same group names for the conformer groups in different reaction networks and keep the groups distinct (as they are now subgroups).
- Ensure mass is conserved option
-
Ensure that all reaction stages (which correspond to sibling groups) have the same mass, and post an error if they do not. This allows checking that mass is conserved throughout the network. You can construct networks in which structures are introduced or removed at any stage; in this case this option should not be selected.
- Specify a representation of haptic ligands option and menu
-
Choose a bond representation of ligands in haptic complexes, from:
-
- Bond to Centroid—Metal center is bonded to a dummy atom at the center of a haptic ligand.
- Bonds to Atoms—Metal center is bonded to all atoms in the haptic ligand.
-
Only available if a haptic complex is added as a conformer group.
- Run button
-
Create and annotate the structures and save them in their conformer groups inside the parent group in the Project Table.
- Status bar
-
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel.If you can submit a job from the panel, the status bar displays information about the current job settings and status for the panel. The settings include the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
The status bar also contains the Help button
 , which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.
, which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.