Reaction Network Profiler Panel
To open this panel: click the Tasks button and browse to Materials → Quantum Mechanics → Reaction Network → Reaction Network Profiler Calculations.
The following licenses are required to use this panel: MS Maestro, OPLS (optional), Jaguar, MacroModel (optional), MS Force Field Applications (optional)
- Using
- Features
- Additional Resources
Using the Reaction Network Profiler Panel
This panel is used to run Jaguar calculations on structures that form parts of a reaction network. The structures must be prepared in the Create Reaction Network Profiler Input Structures Panel, which annotates the structures with information on restraints for conformational search and preoptimization of transition states, as well as tagging structures that are transition states. There is no restriction on the number of structures or the type of structures, so you could process several reactions at the same time. However, the output is returned in a single entry group named with the job name. This entry group contains entry groups of the optimized structures that match the entry groups of the structures that are selected as input. You can view a reaction profile in the Reaction Profile Viewer Panel.
If subjobs fail for some structures, you can import these structures from the jobname_failed.maegz file. Each structure has a set of Successful step-name Boolean properties, which you can use to determine which steps in the workflow failed. As these are secondary properties you will have to show them in the Project Table first—see Organizing Properties for more information.
The structure output file is named jobname-out_rxnwf.mae, and contains the geometries of the conformers generated at the critical points. The geometries are the optimized or transition state geometries. It also contains energy properties from all of the given stages, including analysis stages. These properties have a stageN suffix, to identify the stage in the network.
In addition to the structure output, there are other output files, jobname_conf_avg_rel_reactants.csv and jobname_conf_avg_rel_parents.json, that contain conformationally averaged energies relative to reactants and relative to parents for points along the reaction path. If multiple temperatures and pressures are specified, the reaction rate for each temperature and pressure is written to a CSV file, jobname_conf_avg_rate_constants.csv.
The panel calculates turn over frequencies using the energetic span model by default. The output file jobname_tof.csv reports both the sum-over-states turn over frequencies and the determining states approximated turn over frequencies, along with information on the energy type, whether conformationally averaged energies or the lowest conformer energy were used, reaction energy, and the determining intermediate, TS, and energy span.
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
Reaction Network Profiler Panel Features
- Import button
- Ensure mass is conserved on import option
- Deduplicate structures using this RMSD option and text box
- Conformational Search tab
- Thermochemistry tab
- Job toolbar
- Status bar
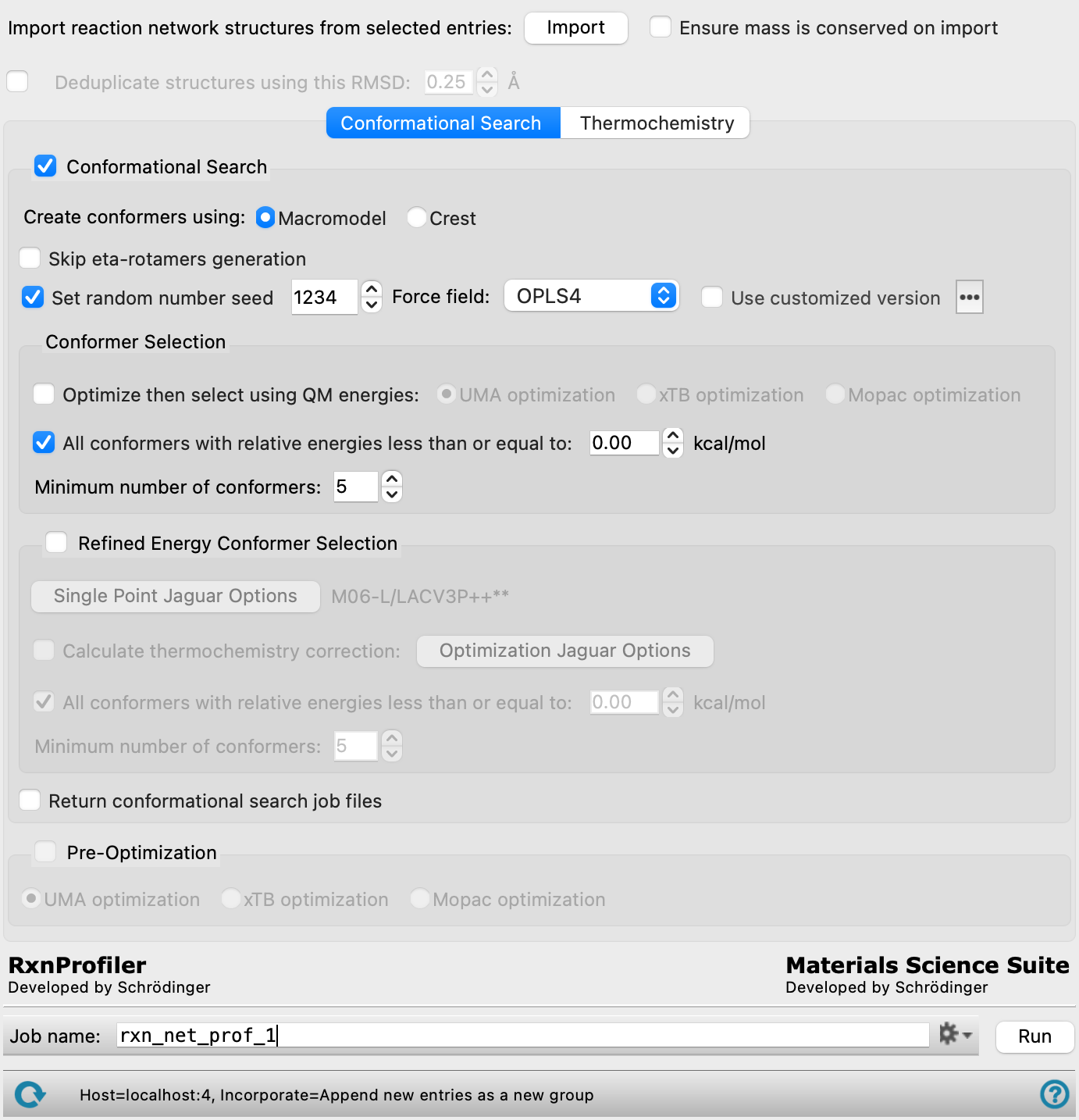
- Import button
-
Import the reaction network structures from the entries that are selected in the Project Table. You should select entry groups that were generated with the Create Reaction Network Profiler Input Structures Panel, as these structures have the restraints specified that will assist in finding the transition states or restrict the conformational search to the desired parts of the structure.
This button is not available if you have added jobs to the network, by clicking the Open button and using the QM Multistage Workflow Panel. To import structures again, you must reset the panel to start a new reaction network.
- Ensure mass is conserved on import option
-
Ensure that all reaction steps in the imported network have the same mass, and post an error if they do not. This allows checking that mass is conserved throughout the network. You can construct networks in which structures are introduced or removed at any step; in this case this option should not be selected.
- Deduplicate structures using this RMSD option and text box
-
Conformers within the specified RMSD threshold are grouped together and assumed to have identical results. Calculations are performed for one representative conformer, and the results copied over to the other conformers in the group. This reduces the number of Jaguar calculations needed.
- Conformational Search tab
-
- Conformational Search option and section
-
Select this option to perform a conformational search, and make settings for the search with the features below. Atom position restraints for the conformational search can be specified in the Create Reaction Network Profiler Input Structures Panel. This option is useful if you want to investigate conformational dependence of a part of the structure that is not involved in the reaction, for example to allow for ligand or catalyst conformations around a known intermediate or transition state geometry. For conformations that may be involved in the reaction, you can pregenerate the conformers and supply them as input to the Create Reaction Network Profiler Input Structures Panel.
- Create conformers using options
-
Choose the tool to use for generating conformers.
-
MacroModel—Use MacroModel to perform a Monte Carlo Multiple Minimum (MCMM) search with 100 steps and the requested minimum or maximum number of structures, using the selected force field. Conformers are ordered by energy, and the thresholds on the number of structures and the energy are applied to this list. Structures within 0.1 Å RMSD of each other are considered the same conformer.
-
Crest—Use the Conformer-Rotamer Ensemble Sampling Tool to perform very efficient conformational space sampling based on metadynamics MD with the GFN2-xTB quantum mechanical method.
The options below are only available if MacroModel was chosen for conformational search.
-
- Skip eta-rotamer generation option
-
Do not generate rotamers for eta-bonded ligands.
- Set random number seed option and text box
-
Select this option to specify a random seed for the conformational search. Specifying the seed allows you to reproduce the results, unless other factors affect them. If this option is not selected, a seed is chosen at random. The value must be between 1 and 78593, inclusive.
- Force field option menu
-
Choose the force field for the conformational search.
- Use customized version option
-
Use your customized version of the OPLS4 force field, rather than the standard version in the distribution.
If the customized version is missing or invalid, the text of this option turns orange and an orange warning icon is displayed to the right, with a tooltip about the problem.
- Parameter set button
-
Select the set of custom parameters for the OPLS4 force field. Opens the Set Custom Parameters Location Dialog Box.
- Conformer Selection section
-
Set options for selection of conformers. This section is not available if you deselect the Conformational Search option.
- Optimize then select using QM energies options
-
Optimize the conformers with the selected method, either UMA, xTB, or Mopac, then select the conformers on the basis of the QM energy using the parameters below.
- All conformers with relative energies less than or equal to option and text box
-
Keep all conformers whose relative energies are at or below the specified threshold value. When selected, the text box for the number of conformers specifies the minimum number. The minimum number is always kept, regardless of the threshold on the relative energies.
- Minimum number of conformers text box
-
Specify the limit on the number of low-energy conformers to keep. The limit depends on whether the All conformers with relative energies less than or equal to option is selected: if it is, you can specify the minimum number of conformers; if it is not, you can specify the maximum number of conformers. The number is counted from the lowest-energy conformer.
The conformational search may find fewer conformers than the specified number, so the value you specify does not guarantee that you will get the minimum or maximum number.
- Refined Energy Conformer Selection option and section
-
Optionally, select this option to select conformers from refined single point energies. This section is not available if you deselect the Conformational Search option.
- Options button
-
Set Jaguar options for the single point energy calculations. Machine learning force fields can be used in place of the quantum mechanics calculations. Opens the Jaguar Options - Reaction Workflow Dialog Box. This dialog box also allows you to specify additional Jaguar keywords. The solvent (if any), level of theory, and basis set are shown to the right of the button.
- Calculate thermochemistry correction option
-
Select this option to include thermochemical corrections to the refined energies.
- Options button
-
Set Jaguar options for the optimization calculations. These are needed to get the frequencies and other thermochemical properties. Machine learning force fields can be used in place of the quantum mechanics calculations. Opens the Jaguar Options - Reaction Workflow Dialog Box. This dialog box also allows you to specify additional Jaguar keywords. The solvent (if any), level of theory, and basis set are shown to the right of the button.
- All conformers with relative energies less than or equal to option and text box
-
Keep all conformers whose relative energies are at or below the specified threshold value. When selected, the text box for the number of conformers specifies the minimum number. The minimum number is always kept, regardless of the threshold on the relative energies.
- Minimum number of conformers text box
-
Specify the limit on the number of low-energy conformers to keep. The limit depends on whether the All conformers with relative energies less than or equal to option is selected: if it is, you can specify the minimum number of conformers; if it is not, you can specify the maximum number of conformers. The number is counted from the lowest-energy conformer.
The conformational search may find fewer conformers than the specified number, so the value you specify does not guarantee that you will get the minimum or maximum number.
- Return conformational search job files option
-
Return the job files for the conformational search to the job directory on the launch host. By default the files are not returned. If the job is large, copying back these files could take some time. Failed jobs are automatically copied back regardless of this setting.
- Pre-Optimization option and section
-
Choose whether you want to pre-optimize an input set of conformer structures with the UMA, xTB, or Mopac methods. This section is not available if you choose Conformational Search.
- Thermochemistry tab
-
- Jaguar section
-
Specify settings for Jaguar optimization, transition state searches, and thermochemistry.
- Options button
-
Set Jaguar options for the optimizations and transition state searches. Machine learning force fields can be used in place of the quantum mechanics calculations. Opens the Jaguar Options - Reaction Workflow Dialog Box. This dialog box also allows you to specify additional Jaguar keywords. The solvent (if any), level of theory, and basis set are shown to the right of the button.
-
Note: You cannot specify the molecular charge or multiplicity with these keywords. They must be specified in the structures themselves, as there is no guarantee that these properties are the same for all structures submitted for optimization from this panel.
- Initial Hessian for transition state searches option menu
-
Specify the Hessian used in transition state searches from the following:
- Schlegal guess
- Fischer-Almolf guess
- Unit Hessian
- Quantum-mechanical
- GFN2-xTB
- Tolerate negative (imaginary) frequencies greater than W wavenumbers text box
-
Consider structures to be at a minimum if they have negative values of the frequency that are above the negative-valued cutoff given in the text box. (The negative values of the frequency are a convenient way of representing the imaginary values in terms of a real number: these represent negative eigenvalues of the mass-weighted Hessian.)
- Temperatures text boxes
-
Specify a range of temperatures for the reaction, by setting the start temperature, the temperature step, and the number of points. The default is one point at 298.15K.
- Pressures text boxes
-
Specify a range of pressures for the reaction, by setting the start pressure, the pressure step, and the number of points. The default is one point at 1 atm.
- Anharmonic Workflow option and section
-
Introduce anharmonicity for low-energy modes to improve the vibrational partition function and thus the rate constants. Each low-frequency normal mode is sampled with single-point calculations to evaluate the anharmonic corrections. The anharmonic curve is determined by a quartic fit to the points along the curve, following Beste [31].
- Applies to normal modes with frequencies less than W wavenumbers text box
-
Specify the cutoff on normal mode frequencies for which anharmonic corrections are computed. Any normal mode with a frequency less than this cutoff is included in the anharmonic correction calculations.
- Factors for scanning normal mode displacements text boxes
-
Specify a grid of points in terms of fractions of the normal mode coordinate for which single-point calculations are run for the anharmonic corrections. The grid is specified by a start fraction, a step value, and the number of points. Steps are taken in both directions from the minimum, so the number of points in the text box is the number in each direction; the total number of points is twice this number.
- Rate constants option
-
Calculate the rate constants for each temperature and pressure, and report them in a CSV file named
jobname_conf_avg_rate_constants.csv.- Transition state theory analysis options
-
Select an option for the TST theory approach used to generate rate constants.
- Standard—Use the standard theory with harmonic frequencies and Boltzmann averaging.
- Extended—Run multiple rate calculations, with various energies, averaging over conformers, harmonic or anharmonic frequencies.
- Return Jaguar job files option
-
Return the job files for the Jaguar jobs to the job directory on the launch host. By default the files are not returned. If the job is large, copying back these files could take some time. Failed jobs are automatically copied back regardless of this setting.
- Specify additional jobs using a Jaguar multistage workflow button
-
Click the Open button to open the QM Multistage Workflow Panel with the reaction network loaded as the first stage (noneditable). You can then add stages to the workflow, including an analysis stage. Clicking Save in the QM Multistage Workflow Panel writes out the updated workflow, which you can then run from this panel. Once you set up a multistage workflow, the Import button is no longer available, as the job is regarded as completely defined. This means you should set up the reaction worfklow first. To import structures again, reset the panel from the Settings button menu.
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The settings button menu has a Reset item, to reset the panel to its default settings.
The Job Settings button opens the Reaction Network Profiler - Job Settings Dialog Box, where you can make settings for running the job.
- Status bar
-
The status bar displays information about the current job settings and status for the panel. The settings includes the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
Use the Reset button
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel. The status bar also contains the Help button
 , which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a
, which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a  button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.
button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.