Reaction Path Interpolation Panel
Create a linear synchronous transit reaction path by interpolation between a reactant structure and a product structure. The path is returned as a set of structures with a property that defines the distance along the path.
To open this panel, click the Tasks button and browse to Materials → Structure Builders → Reaction Path Interpolation.
The following licenses are required to use this panel: MS Maestro
- Overview
- Using
- Features
- Additional Resources
Overview of Reaction Path Interpolation
The reaction path interpolation procedure is based on the work of Halgren and Lipscomb[1].
The algorithm proceeds by performing a linear interpolation of the coordinates between the reactants and the products, to produce a set of target coordinates. Interpolation of internal and distance coordinates do not, however, produce a consistent set of Cartesian coordinates, so a Cartesian interpolation is also done as a starting guess, and these Cartesian coordinates are adjusted in a nonlinear least-squares procedure to minimize the difference between the internal or distance coordinates generated from the Cartesian coordinates and those determined from the interpolation.
The orientation of the structures is important for the Cartesian interpolation. Before the interpolation, begins, the product structure is superimposed onto the reactant structure, using the nonreactive (or least reactive) atoms to perform the superposition. This procedure minimizes the changes in the coordinates. It also prevents the lines between corresponding atoms (on which the linear Cartesian interpolation is done) from passing through the same point.
The structure obtained at each step of the interpolation is also superimposed on the reactant structure. This process helps prevent drift (translation or rotation of the entire structure), and also makes visualization of the path easier.
Using the Reaction Path Interpolation Panel
In general, the reaction and product structures should be constructed so that the structures are pre-positioned for reaction (in either direction) in a pre-reactive or post-reactive complex. Structures that are pre-positioned close to the transition state provide better reaction paths than structures that are at their equilibrium geometries and far from the transition state. It is highly recommended that you build the product structures by modifying the reactant structures, breaking bonds and forming new bonds as necessary but not deleting any atoms. Keeping all the atoms is critical to maintaining the atom ordering, which is necessary for constructing the path. The atom numbering must match, otherwise the reaction path will be meaningless. To ensure that no automatic deletion of hydrogens is done, deselect Allow united atom types while building and Adjust number of hydrogens following additive build operations under Behavior settings in the Builder Preferences tab of the Preferences Panel.
If you build the products from the reactants, you should do some kind of restrained minimization of the structures that relieves strain but does not significantly reposition the product structures.
If you have structures that do not fulfil these conditions, the panel offers you ways to change the atom numbering and position the reactants and products, as explained below.
For certain reaction types, you can select Pre-form reaction complex if possible to form a complex of the reactants and a complex of the products, to assist in defining the path. The reactive atoms are identified and the structures are positioned with respect to each other to form a pre-reactive or post-reactive complex. This is only available for bimolecular reactions in which transfer of atoms happens only from one molecule to the other (not exchange of groups), such as SN2 reactions, donation of a group to another molecule, or reaction of two molecules to form a single molecule (and its reverse, unimolecular decomposition).
Otherwise, you could consider using the Probe Grid Scan Panel to pre-position the reactants and the products. If you do, you will probably need to renumber the atoms in the product complex to match the reactant complex.
You can renumber the atoms if the reactant and product numbering schemes do not match, in the Reorder Atoms panel. This panel shows a 2D image of the reactant and product complexes, and allows you to visually select the corresponding atoms for renumbering.
The points along the reaction path can be chosen as an evenly-spaced set or as a list of points. The path is defined with a value of 0 for the reactants and 1 for the products. You can add points around the approximate location of the transition state, spaced at 0.02 in the path coordinate with 5 points on each side, by selecting Add extra points clustered in transition state region. The presumed transition state location can be chosen as early (25%), midway (50%) or late (75%) in the reaction path. Any existing points that fall in the range covered by the added points are removed.
The simplest linear synchronous transit path can be obtained by interpolating the Cartesian coordinates of the corresponding atoms in the reactants and the products. This simple path could involve clashes if the structures were not properly aligned - for example, if the lines between the corresponding atoms all passed close to the same point. Distance and internal coordinates are over-determined sets, so fitting is required. Constraints are imposed so that a smooth change in the coordinates between the reactant and product structures is obtained.
To obtain an initial reaction path for the optimization, the product structure is superimposed onto the reactant structure before the interpolation begins, using the nonreactive (or least reactive) atoms to perform the superposition. The structure obtained at each step of the interpolation is also superimposed on the reactant structure. This process helps prevent drift (translation or rotation of the entire structure), and also makes visualization of the path easier.
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
When the path has been generated, the output Maestro file contains all of the structures: first the reactants, then the reactant complex (if requested), the steps in the path, the product complex (if requested), then the products. The step index and distance along the path (in the range 0 to 1) are included as entry properties, and the reactive atoms are identified with an atom-level property. To see these properties in the Project Table, you will have to choose to show them explicitly, or show all properties, as they are not shown by default.
Reaction Path Interpolation Panel Features
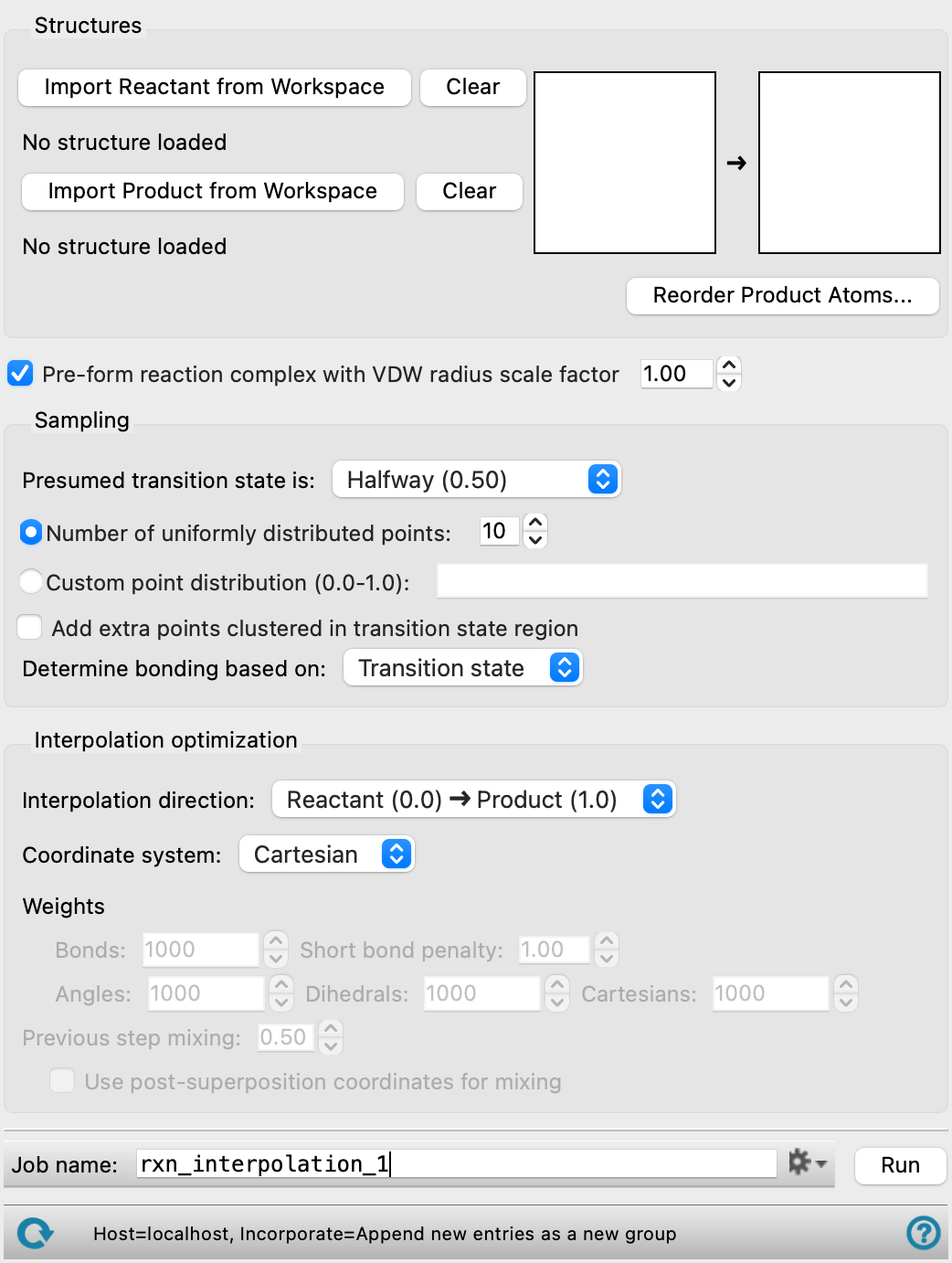
Structures section
In this section you define the reactant and product structures, renumbering the product atoms if necessary. The structures must have the same elemental composition.
- Import Reactant from Workspace button
-
Place the reactant or reactants in the Workspace, positioned for the reaction, then click this button to identify the contents of the Workspace as the reactant structure. A 2D sketch of the reactants is displayed in the reaction diagram. This 2D sketch does not necessarily represent the 3D geometry or stereochemistry.
- Import Product from Workspace button
-
Place the product or products in the Workspace, positioned for the reaction, then click this button to identify the contents of the Workspace as the product structure. A 2D sketch of the products is displayed in the reaction diagram.
- Clear buttons
-
Clear the reactant structure or the product structure from the workflow. The 2D sketch is removed from the reaction diagram.
- Reaction diagram
-
This diagram shows a 2D sketch of the reactant structure and the product structure.
- Reorder Product Atoms button
-
If the reactant and product structures do not have the same atom numbering, click this button to open the Reorder Atoms Dialog Box and change the numbering of the product atoms. The atom numbering must match, otherwise the reaction path will be meaningless.
In the dialog box, the reactants constitute the reference structure and the products constitute the comparison structure (the one that is reordered).
Sampling section
In this section you specify the set of points along the reaction path that you want to calculate.
- Presumed transition state is option menu
-
Choose the approximate location of the transition state along the path from this option menu. This location is used when adding points to the path and when determining the bonding.
- Number of uniformly distributed points option and menu
-
Select this option if you want a uniform distribution of points along the reaction path, and specify the number of points. These points do not include the reactant and product structures, so if you choose 10 points, 12 structures are returned: the reactant, the product, and the structures for each point.
- Custom point distribution text box
-
Specify the distribution of points along the reaction path as a space-separated list of values in the range between 0.0 and 1.0. The values 0.0 and 1.0 are reserved for the reactants and the products.
- Add extra points clustered in transition state region option
-
Add 10 points to the path around the presumed transition state with a step size of 0.02.These points replace any points specified by the uniform or custom distribution that lie within the range of the extra points.
- Determine bonding based on option menu
-
Specify how bonds (connectivity) are assigned in the interpolated structures.
- Reactant—Use the connectivity from the reactant for all structures
- Product—Use the connectivity from the products for all structures.
- Transition state—Change the connectivity from that of the reactant to that of the product along the reaction path, with the change occurring in the region of the presumed transition state. In this region, bonds that are broken or formed are represented as zero-order bonds.
- Interpolation direction option menu
-
Use this option menu to set the direction in which the interpolation proceeds: from reactant to product or from product to reactant. For a simple Cartesian interpolation, the path should be independent of the direction, but for other interpolations, the path may vary with the direction if step mixing is used.
Reaction complex section
Pre-position the reactant molecules for the reaction, and the product molecules for the reverse reaction. Structures that are pre-positioned close to the transition state provide better reaction paths than structures that are at their equilibrium geometries and far from the transition state.
This feature is limited to unimolecular decompositions that form two molecules and bimolecular combination reactions that do not involve exchange of atoms (any leaving group comes from only one of the original molecules) So, for example, an SN2 reaction would work as the nucleophile combines with the substrate and the leaving group comes from the substrate, but a hydrolysis reaction would not work because the water donates a hydrogen and accepts another fragment.
- Pre-form reaction complex if possible option
-
Place the reactant structures into a complex that is pre-positioned for reaction, and similarly place the product structures into a complex. This is done with rigid-body movement of the molecules.
- VDW radius scale factor text box
-
When positioning the molecules in the complex, ensure that the intermolecular distances are no smaller than the sums of the scaled radii of the molecules involved. A larger scaling factor places the molecules further apart. The molecular diameter is taken to be the largest atom-atom distance plus the van der Waals radii of the atoms involved; the molecular radius is half this value.
Interpolation optimization section
Set parameters for the optimization of the structures along the path to bring them as close as possible to the linear synchronous path while avoiding unreasonable structures.
- Coordinate system option menu
-
Choose the coordinate system in which the interpolation is done.
- Internal—Use the internal coordinates of the structures (bond distances, bond angles, dihedrals). This is a set of redundant internal coordinates that includes the union of the coordinate sets for both products and reactants. This option can be used to avoid some kinds of atomic collisions along the path, and works for the greatest number of reactions.
- Distance—Use the set of all interatomic distances. This option can help avoid over-estimation of bond lengths.
- Cartesian—Use the set of Cartesian atomic coordinates. This is the fastest option
The choice of a coordinate system that is interpolated is critical to finding a suitable reaction path. Cartesian coordinates may be suitable for some reactions, such as the classic SN2 reaction, X− + CH3Y → CH3X + Y− (if the halides X and Y and the carbon atom remain in the same order). The rearrangement of HCN to HNC cannot be done with Cartesian interpolation, however, as the hydrogen would have to pass through both heavy atoms. A better coordinate system for this reaction is the set of distance coordinates: the HC distance has to increase in a linear interpolation, which forces the hydrogen to go around the CN rather than through.
- Weights text boxes
-
Set weight factors for the squared coordinate differences in the least squares procedure used in fitting the reaction path. These weights can be set in the Bonds, Angles, Dihedrals, and Cartesians boxes. The weights that are available depend on the coordinate type. Increasing the value increases the rigidity of the interpolated coordinates (damps changes). If convergence difficulties are encountered, changing the weights can help. Increasing or decreasing the Cartesian weight can remedy troublesome reaction paths more effectively than changing the other weighting terms. When optimizing on distances, each distance is considered a "bond" for weighting purposes.
- Short bond penalty text box
-
Apply the specified penalty for bonds that are too short. This value can be increased if the reaction path has atom-atom collisions. The penalty is applied in the form of penalty/R, where R is the bond distance. This penalty is available when using internal or distance coordinates.
- Previous step mixing text box
-
Specify the amount of the optimized Cartesian coordinates from the previous step to mix into the interpolated guess for the current step. The resultant coordinates form the guess for the optimization of the current step. Mixing can help to avoid collisions and to create continuous reaction paths.
- Use post-superposition coordinates for mixing
-
Use the coordinates from the previous step after superposition on the reactant structure. If this option is not selected, the optimized coordinates from the previous step before superposition are used. Only available if the mixing of the previous step is nonzero.
Job toolbar
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Reaction Path Interpolation - Job Settings Dialog Box, where you can make settings for running the job.
Status bar
The status bar displays information about the current job settings and status for the panel. The settings includes the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
Use the Reset button  to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel.
The status bar also contains the Help button  , which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a
, which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a  button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.
button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.