Nanoreactor Panel
Sample potential energy surfaces or determine elementary reaction networks using xTB metadynamics and optionally identify their thermodynamic sorting with free energy refinement.
To display this panel: click the Tasks button and browse to Materials → Quantum Mechanics → Reaction Network → Nanoreactor Calculations
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
The following licenses are required to use this panel: MS Maestro, MS Reactivity, Jaguar, MS Force Field Applications (optional)
- Using
- Features
- Additional Resources
Using the Nanoreactor Panel
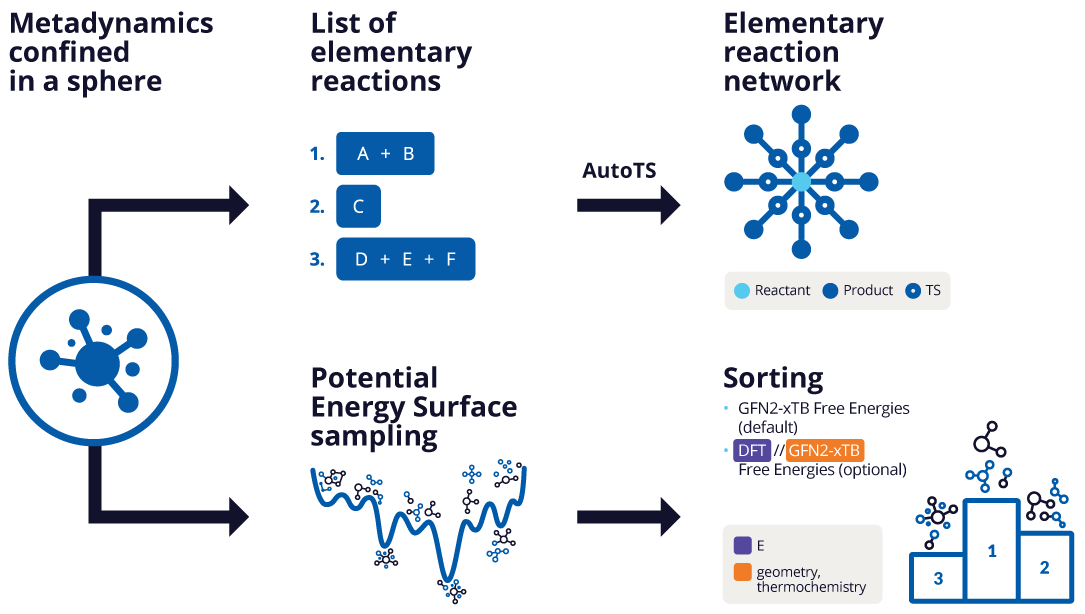
The Nanoreactor Panel can be used in two ways (1) to explore thermal and chemical product decomposition space through the identification of minima states on the potential energy surface and (2) identify relevant elementary reactions from a structure that is a local minimum on the potential energy surface. This panel provides insight into decomposition products without requiring user knowledge of the outcome. The input to the panel must be a single entry, but this entry can contain several molecules, ions, or radicals, but not isotopes. The nanoreactor workflow consists of the following steps:
-
The input structure’s geometry is optimized with the GFN2-xTB semiempirical tight-binding method using the charge, initial spin state, and optional implicit solvent specified. Related files can be found in the job directory under
input_opt. -
The optimized structure undergoes multiple replicates consisting of xTB molecular dynamics, xTB metadynamics, and xTB optimization [58].
-
Molecular dynamics—A constrained molecular dynamics simulation is run using xTB on the optimized input structure at the specified initial spin state. The optimized structure is confined in a non-periodic sphere represented by a spherical wall potential. The specified parameters are used for the relaxation protocol. Related files can be found in the job directory under
replicate_n/md. -
Metadynamics—A constrained root-mean-square-deviation (RMSD) based metadynamics simulation is run to automatically sample thermal and chemical product decomposition space at the specified initial spin state. Related files can be found in the job directory under
replicate_n/metadynamics. -
Geometry optimization—A GFN2-xTB geometry optimization is performed on each frame of the molecular dynamics and metadynamics simulations at the specified final spin state. The xTB free energy at 298.15 K and 1 atm is reported for each frame. For potential energy surface sampling, related files can be found in the job directory under
replicate_n/frame_opt.tar.gz.
-
-
The xTB optimized reactant(s), all frames from the molecular dynamics simulation, and all frames from the metadynamics trajectories (which contain the decomposition products) are combined. For potential energy surface sampling, the frequency of each frame is calculated as the number of frames that have the same SMILES string. The lowest energy frame of each unique structure is retained. For elementary reaction network calculations, frames that are not the same as the reactant structure are retained. The structures can be further filtered based on energy if specified.
-
Corrected free energies can be optionally calculated for each unique structure: electronic energy is calculated with DFT (Jaguar) and thermal corrections are calculated with xTB. The reactant is considered at the initial spin state and all other unique structures are considered at the final spin state for potential energy surface sampling. Refinement with DFT provides high-fidelity thermodynamic sorting of all nanoreactor products and is recommended. Please note that calculation time increases significantly for larger structures. Related files can be found in the job directory under
refinement.tar.gz
To visualize the results, you can use the Nanoreactor Viewer Panel (click the Tasks button and browse to Materials → Quantum Mechanics → Reaction Network → Nanoreactor Results). The viewer panel allows you to view output structures based on relative energies.
Nanoreactor is an integration and extension of Grimme's implementation. Please cite the Grimme reference and others [58, 88, 89] in any publication that contains results from the use of this panel.
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button and choose Write.
Nanoreactor Panel Features
- Use structures from option menu
- Open Project Table button
- Calculation Type options
- Charge text box
- Number of unpaired electrons for initial state text box
- Final state text box
- Solvent option menu
- Relaxation time text box
- Metadynamics time text box
- Trajectory frame interval text box
- Temperature text box
- Scaling factor for cavity radius text box
- Biasing Potential section
- Replicates text box
- Remove high energy products based on xTB Free Energy option and text box
- Refine final energy option and section
- Return trajectory files option
- Job toolbar
- Status bar
- Use structures from option menu
-
Choose the structure source for the current task.
- Workspace (included entry)—Use the entry that is currently included in the Workspace. Only one entry must be included in the Workspace.
- Workspace (included entry)—Use the entry that is currently included in the Workspace. Only one entry must be included in the Workspace.
- Open Project Table button
-
Open the Project Table panel, so you can

- Calculation Type options
-
Specify the type of nanoreactor calculation to run on the system:
- Potential energy surface sampling—Systematically explore and rank minima states on potential energy surfaces. See Using the Nanoreactor Panel.
- Elementary reaction network—Identify relevant elementary reactions from a structure that is a local minimum on the potential energy surface. This mode produces products that may be used as inputs for transition state searching using tools such as the AutoTS: Perform Calculations Panel. Not all products generated will correspond to true elementary steps, this should be confirmed through transition state analysis and subsequent intrinsic reaction coordinate (IRC) calculations using the AutoTS: Perform Calculations Panel.
- Charge text box
-
Specify the overall charge of the system. Negative, positive, and zero values are allowed.
- Number of unpaired electrons for initial state text box
-
Specify the number of unpaired electrons in the system at its initial state. The inital xTB optimization, molecular dynamics, and metadynamics are performed at this spin state.
- Final state text box
-
Specify the number of unpaired electrons in the system at its final state. The xTB geometry optimizations and optional DFT electronic energies that follow the metadynamics simulations are calculated at this spin state.
-
This parameter can be set to the same or a different value as the Initial state unpaired electrons parameter. A differing value is helpful here when your products have a different spin state than the reactants.
- Solvent option menu
-
Choose a solvent to use for the nanoreactor calculation from this menu. By default, the calculation runs in the gas phase. If a solvent is selected, the Poisson-Boltzmann solvent model is used. Note that energy refinement is calculated in the gas phase regardless of solvent choice.
- Relaxation time text box
-
Specify the desired relaxation time for the molecular dynamics simulation in ps. xTB MD is run on a system consisting of the input structure, which has been geometry optimized with GFN2-xTB, without periodic boundary conditions.
- Metadynamics time text box
-
Specify the desired simulation time for metadynamics in ps.
- Trajectory frame interval text box
-
Set the interval for saving points on the molecular dynamics and metadynamics trajectories, in fs. This is the amount of time between frames in the trajectory. Only present when Elementary reaction network is selected for the Calculation Type.
- Temperature text box
-
Specify the temperature to be used for molecular dynamics and metadynamics simulations, in Kelvin.
- Scaling factor for cavity radius text box
-
Specify the scaling factor to use for the radius of the repulsive potential cavity used to confine the system for molecular dynamics and metadynamics simulations. The radius of the spherical wall potential is determined by the largest interatomic distance in theinitial GFN2-xTB optimized structure multiplied by the specified scaling factor.
- Biasing Potential section
-
Set parameters that influence the biasing potential.
- Strength options
-
Specify the strength of the biasing potential. Higher kpush values result in more reactions but can lead to non-physical structures if too high:
- Automatic—The strength of the biasing potential will be set to 0.02 * number of atoms.
- N text box—Specify the strength of the biasing potential.
- Width text box
-
Specify the width of the biasing potential. Higher values of alpha can result in a narrow biasing potential, meaning that the system can avoid the biasing potential with smaller changes to the structure.
- Replicates text box
-
Specify the number of replicates to run. The replicates consist of the xTB molecular dynamics relaxation, metadynamics simulation, and structure optimization for each frame of the metadynamics simulation. See Using the Nanoreactor Panel.
- Remove high energy products based on xTB Free Energy option and text box
-
Specify the relative energy threshold at which products should be removed from the returned unique structures. The relative energy threshold is the xTB free energy relative to the starting material (reactants). The structures that have an energy higher than the specified threshold are filtered out from the workflow replicates. Removing higher energy products can be helpful when lower energy products are of interest.
- Refine final energy option and section
-
Compute the corrected free energy for each unique product of the simulation. The corrected energy is calculated in the gas phase by summing the SCF energy from a DFT calculation using Jaguar and the thermochemistry correction from a GFN2-xTB calculation. The thermochemistry correction is calculated at 298.15 K and 1 atm. This option is not selected by default.
You can use the Jaguar Options button to set additional parameters for the Jaguar calculation.
This option must be selected to view the plots in the Refined Energy and xTB vs Refined Free Energy tabs of the Nanoreactor Viewer Panel.
- Options button
-
Set Jaguar options for the nanoreactor calculation. Machine learning force fields can be used in place of the quantum mechanics calculations. Only available if Refine final energy is selected. Opens the Jaguar Options - Nanoreactor Dialog Box. This dialog box also allows you to specify additional Jaguar keywords. The solvent (if any), level of theory, and basis set are shown to the right of the button.
- Remove high energy products based on refined Free Energy option and text box
-
Specify the relative energy threshold at which products should be removed from the returned refined structures. The relative energy threshold is the corrected free energy relative to the starting material (reactants). The structures that have an energy higher than the specified threshold are filtered out from the results. Removing higher energy products can be helpful when lower energy products are of interest.
Only available if Refine final energy is selected.
- Return trajectory files option
-
Select this option to include trajectory files from the molecular dynamics and metadynamics calculations to the job directory. Note that the amount of data stored in these files can be large. These files contain all of the trajectory data, so you will not be able to view the trajectories for these stages if you do not select this option. Deselecting this option does not affect the use of the Nanoreactor Viewer Panel.
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Nanoreactor - Job Settings Dialog Box, where you can make settings for running the job.
The number of processors requested is divided as equally as possible amongst the number of replicates. For each replicate, the MD and metadynamics stages will run in parallel on up to 8 of the allotted processors if available. Geometry optimizations and energy calculations are distributed across all processors allotted to that replicate as efficiently as possible.
- Status bar
-
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel.If you can submit a job from the panel, the status bar displays information about the current job settings and status for the panel. The settings include the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
The status bar also contains the Help button
 , which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.
, which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.