IFD-MD Panel
Optimize the binding pose of a ligand to a receptor using a template complex with the IFD-MD protocol.
To open this panel: click the Tasks button and browse to Receptor-Based Virtual Screening → IFD-MD. A dialog opens, giving the choice of this panel or the Induced Fit Docking panel: clicking the button opens the panel, There is also an option to remember the choice and open the chosen panel directly in future.
- Overview
- Using
- Features
- Additional Resources
Overview of IFD-MD
Many structure-based drug design (SBDD) methods require accurate, atomic-level detail of the target protein in complex with a ligand in order to perform optimally—including methods such as free energy perturbation (FEP+) theory. Consequently, the domain of applicability of SBDD is limited by the availability of high-resolution crystal structures of protein-ligand complexes. However, even when the exact protein-ligand complex structure is not available, highly similar ones may be. IFD-MD starts with the available structure and predicts the atomic details of the protein-ligand complex structure needed for SBDD. It is capable of confidently predicting the binding pose of one ligand, starting from a structure of the target protein with a very different bound ligand. When combined with Prime Homology Modeling, IFD-MD can even predict a target protein-ligand complex structure without any structures of the target protein, by using a structure of a highly homologous protein as a starting protein.
Often the only differences in protein structure between the available structure and the one that is needed for SBDD are the movements of a couple of side chains or a small loop motion. These small changes can, however, have a large impact on the accessible ligand binding modes. IFD-MD uses a combination of docking algorithms, water thermodynamics, empirical scoring functions, implicit solvent force field energies, and explicit solvent metadynamics trajectories to explore the motions of the target protein and simultaneously determine their relative energetics. This technology makes it possible to create accurate “first looks” at the protein-ligand interactions for novel active compounds before a crystal structure is solved, and even allows for accurate structures of protein-ligand binding when starting from homology models.
Overview of Consensus IFD-MD
Consensus IFD-MD assumes that congeneric ligands share one binding mode in the protein. Utilizing this assumption, consensus IFD-MD independently docks two to three congeneric ligands into an ensemble of protein structures and only returns predicted binding modes that satisfy this assumption.
Employing ensemble receptor structures ensures coverage of large receptor conformational changes, it can also result in a substantial increase in the computational cost for IFD-MD. To mitigate the additional computational burden, the IFD-MD metadynamics step was skipped for computational efficiency resulting in approximately 1/10 of the GPU resource usage compared to full IFD-MD. The IFD-MD metadynamics step is only used for scoring in IFD-MD meaning that its exclusion does not affect the quality of the poses returned, only their ranking. The use of the consensus binding mode assumption compensates for this reduced scoring accuracy by discarding poses that score competitively but not consistently across the congeneric series. In an example for modeling hERG inhibition, even with the docking of 3 ligands, the computational cost of docking them into 3 hERG receptor conformation is roughly equivalent to the GPU cost of running 90% of a single full IFD-MD job, significantly reducing the overall cost compared to running 9 full IFD-MD jobs.
Consensus Binding Mode Analysis (CBMA) finds and clusters common binding modes among the docked congeneric ligands from all the independently run IFD-MD jobs. The clustering is performed based on the Maximum Common Substructure (MCS) ligand RMSD, ensuring that the poses with similar binding modes are grouped together.
Using the IFD-MD Panel
To run an IFD-MD job requires specification of a template complex and the ligand for which the pose prediction is desired. The results of the job are a set of poses in a Maestro file: each "pose" is a ligand-receptor complex. The primary Maestro property generated by this panel is the IFD-MD Score. Other properties for contributions to the score are added as secondary properties. The IFD-MD Score is only useful for the relative ranking of poses: it cannot be used to compare poses across different IFD-MD runs, for example, where the input ligand or receptor are different.
Poses with similar IFD-MD scores might warrant further investigation. For example, if the top three poses all had scores within 2.5 units of each other, it might be worthwhile to examine these three poses further, such as by running FEP+ and comparing with known SAR data.
For proteins that do not have a crystal structure, you can use FEP+ in combination with Prime Homology Modeling and IFD-MD to predict the structure of a protein-ligand complex, using the relative binding affinities of a series of congeneric ligands whose experimental binding affinities are known. This can be done by creating multiple Prime homology models with subtle differences in the ligand binding pose and the organization of side chains in the binding site. FEP+ predictions of binding affinities can then be made for the congeneric ligand series using each of the homology models created. A homology model that can correctly predict the binding affinities of these ligands is likely to be structurally accurate in the critical region. This homology model can then be used for a range of structure-based drug design methods, including use of FEP+ to prospectively predict the binding affinities of novel compounds. It is possible that none of the homology models predict the binding affinities of the known ligands, or that the application to unknown ligands do not produce good results due to subtle errors. However, a homology model that predicts the known ligand affinities is likely doing so by reproducing many of the contacts that would be resolved by generating a crystal structure of the protein-ligand complexes.
See IFD-MD Best Practices for recommendations on structure preparation and template selection.
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
Note: To restart an IFD-MD panel job, simply rerun the original command. For reference, the original command used is listed at the top of the IFD-MD logfile next to "Commandline:"
IFD-MD Panel Features
- Docking type options
- Dock into target protein options
- Structures tab
- Reaction tab
- Add distance constraints option
- Calculate force-field customizations for target ligand option
- Add membrane protein template option
- IFD-MD Best Practices link
- Advanced Options link
- Job toolbar
- Status bar
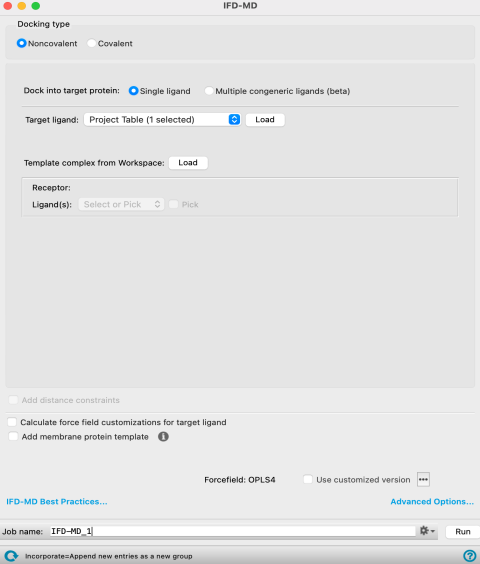
The IFD-MD Panel in Noncovalent Docking configuration.
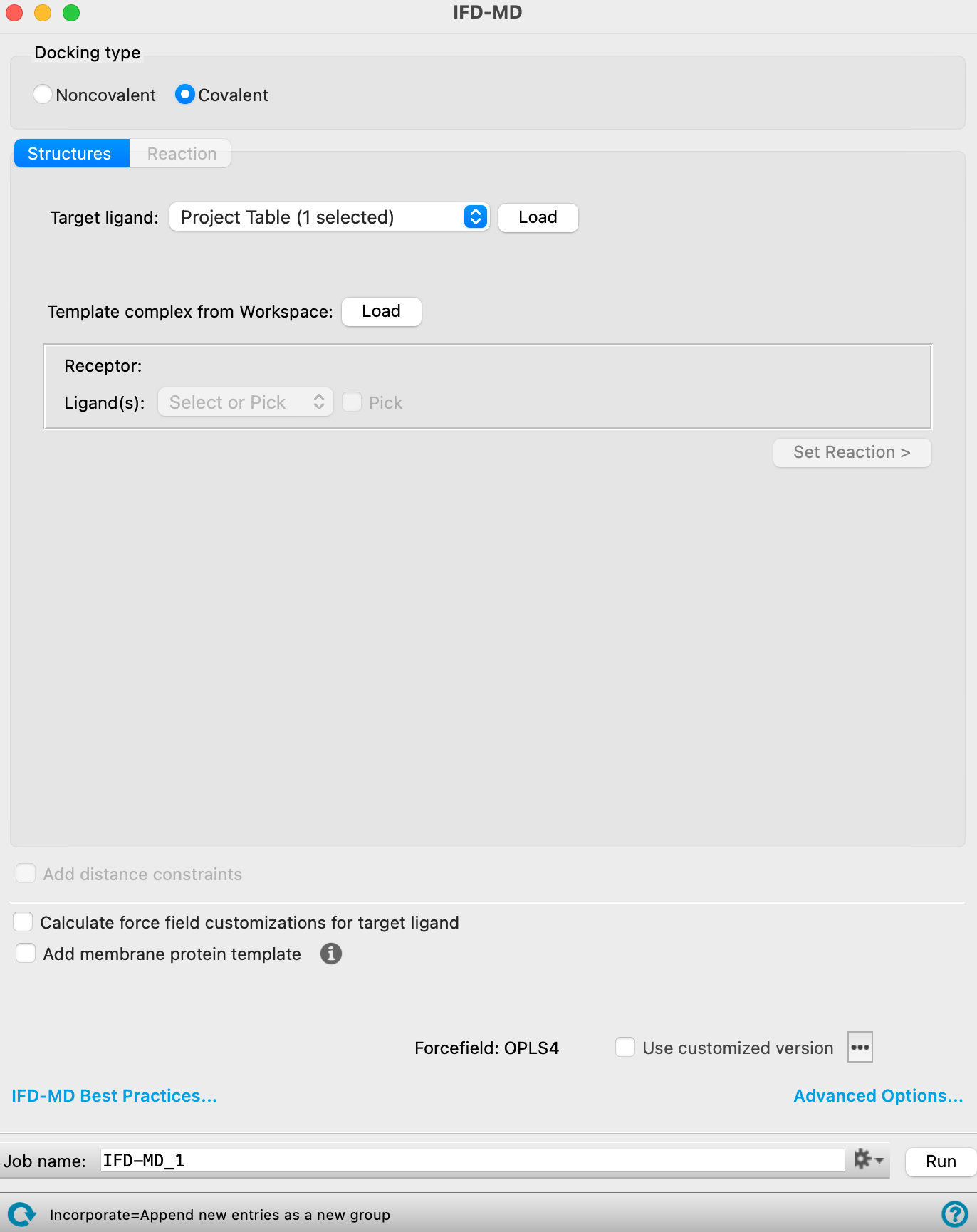
The IFD-MD Panel in Covalent Docking configuration.
- Docking type options
-
Choose the docking method:
Non-covalent—Ligands are docked based on nonbonded interactions (hydrogen bonds, pi-pi interactions, etc.). When you choose this option, only the Dock into target protein options and Structures tab are available.
Covalent—Ligands are docked by forming a covalent bond with the receptor. The covalent bond is defined by a specified reaction. When you choose this option, both the Structures tab and the Reaction tab are available.
- Dock into target protein options
-
Choose the button for either Single ligand, or Multiple congeneric ligands.
If Multiple congenetic ligands is selected, only the Target ligand and the Target protein option menus in the Structures tab are available.
- Structures tab
-
Choose the target ligand, the receptor and the template ligand.
- Target ligand option menu
-
Choose the structure source for the target ligand. You can choose a macrocycle as the ligand; if you do, text is displayed below indicating that macrocycle docking is in effect. If Multiple congeneric ligands was selected for the Dock into target protein option, you will choose two or three ligands instead of a single ligand.
-
Project Table (N selected entries)—Use the entry that is currently selected in the Project Table. Only one entry must be selected. When this option is chosen, the Load button is displayed.
-
File—Use the specified file. When this option is selected, the Browse button is displayed.
-
- Target protein option menu
-
Choose the target protein from the dropdown. The available options are CYP 3A4, CYP 2D6, CYP 2C9, PXR, and hERG. This option is only available if Multiple congeneric ligands was selected for the Dock into target protein option.
- Load button
-
Click this button to load the selected entry into the workflow as the target ligand.
-
A dark exclamation point with tooltip will display if the ligand has torsions that are not modeled in the current force field. In the case of single ligands, the tooltip will state that you will need to check the Calculate force field customizations option to generate the missing parameters before running the IFD-MD job. In the case of multiple ligands, the tooltip will state that custom force field parameters are needed to adequately model the structure. Use the Force Field Builder - OPLS4/OPLS5 Panel, available in the Task Tool, to generate the new parameters before running IFD-MD.
- Browse button
-
Click Browse and navigate to the structure file. The first ligand the file is used; anything else is discarded.
- Template complex from Workspace section
-
In this section, the template receptor-ligand complex and the template ligand that is in the desired binding site are defined.
- Load button
-
Click this button to load the selected entry into the workflow as the template receptor-ligand complex. The text boxes below are populated with the details of the entry that contains the receptor and the ligands in the complex.
- Receptor text box
-
This noneditable text box shows the title of the entry containing the complex to be used in the docking. It is a complex rather than just a receptor, because the ligand in the complex is used to help bias the IFD calculation, hence it must be within the same frame of reference as the binding site, and making all the key contacts. The text box displays an error message if the Workspace does not display a receptor-ligand complex.
- Ligand option menu and Pick option
-
Select the template ligands by checking the box next to the desired ligands, or by selecting the Pick option and picking the ligands in the Workspace. The ligands in the Workspace are automatically detected; however if you pick a ligand in the Workspace that is not on the menu, it is added and checked. The selected ligands are displayed on the top box of the option menu as a comma-separated list; they are also selected in the Workspace.
The ligands you select must all be in the same binding site, and they can be superimposed or clashing. IFD-MD considers all the template ligands for building a pharmacophore model of the binding site. There are some scenarios where this is useful—for example if the target is a promiscuous binder and ligands have been observed docking in a number of different orientations.
For covalent docking, the template ligand can also be covalently bound to the receptor, but non-covalently bound template ligands remain valid as well.
- Reaction tab
-
Choose the reactive residue and reaction type. For detailed information, see the Reaction Type Tab section in the Covalent Docking Panel topic.
- Reactive residue text box and Pick option
-
Specify which residue (or site) in the receptor the target ligand will be bonded to. You can pick the residue in the Workspace by selecting Pick and picking an atom in the desired receptor residue. The residue must be one of those defined by the reaction type—see the table under Reaction type option menu in the Covalent Docking Panel topic for a list of allowed residues. It must also have a standard residue name.
Make sure you do not pick a residue in the ligand, if you have it displayed in the Workspace.
- Reaction type option menu
-
Choose the reaction type from this option menu. This menu is populated by standard reaction types that are possible based on the residue and ligand selected. See the table under Reaction type option menu in the Covalent Docking Panel topic for a list of standard reaction types. If the reaction type that you are interested in is not supplied in the default set, choose Custom (from file) to use a custom reaction type, then import the reaction type definition (
.cdockfile) in the file browser that opens. For instructions on creating a custom reaction type, see Custom Chemistry Definitions for Covalent Docking. - Reactive ligand region (SMARTS) section
-
Once the reaction type has been selected, this section is displayed. The SMARTS pattern for the region is displayed next to the section heading. The ligand is displayed in 2D with the sites that are possible for a reaction highlighted. The reaction site is shown in pink.
- Reaction site option menu
-
If there are multiple reaction sites, choose the site for the reaction from the sites listed on this menu. This menu is not shown if there is only one possible site. The new site is highlighted in pink.
- Chirality option menu
-
If the reaction at the site chosen creates a chiral center, this option menu is displayed so you can set the chirality for the center.
- Add distance constraints option
-
Select to show the table displaying distance constraints. Up to a maximum of four constraints can be added. This option is only available if Single ligand is selected for the Dock into target protein option. Click Add to open a popup to specify the Ligand Atom, Receptor Atom, the Distance, and the Tolerance for each distance constraint. Choose the Ligand atom by clicking the desired atom represented in the popup image. For the Receptor atom, choose by clicking the Pick option and then selecting an atom in the Workspace. For Tolerance and Distance fields, enter a value or use the arrows to adjust the value. To delete a distance constraint, select the corresponding row in the table, then click Remove.
- Calculate force-field customizations for target ligand option
-
Run the force-field builder as part of the IFD-MD job to identify missing torsional parameters in the target ligand and include them in the current job. The new parameters are added to the current customization. Missing torsional parameters will result in a dark exclamation mark appearing with a tooltip. In the case of single ligands, the tooltip will state that you will need to check the Calculate force field customizations option to generate the missing parameters before running the IFD-MD job. In the case of multiple ligands, the tooltip will state that custom force field parameters are needed to adequately model the structure. Use the Force Field Builder - OPLS4/OPLS5 Panel, available in the Task Tool, to generate the new parameters before running IFD-MD.
- Add membrane protein template option
-
Select this option to include an explicit membrane in the workflow. When you select the option, a Browse button is displayed so you can locate a PDB file that contains the membrane (usually from the OPM database). The protein structure in this PDB file should be similar to the template protein (receptor). The template protein is aligned to the membrane protein and the membrane is added.
- IFD-MD Best Practices link
-
Open the IFD-MD Best Practices in your browser.
- Advanced Options link
-
Set advanced options for the IFD-MD docking. Opens the Advanced Options Dialog Box in your browser.
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
- Status bar
-
The status bar displays information about the current job settings and status for the panel. The settings includes the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
Use the Reset button
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel. The status bar also contains the Help button
 , which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a
, which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a  button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.
button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.