Defect Formation Energy Panel
Calculate the defect correction energy for the formation energy of a point defect using the zero defect correction and alignment term.
To display this panel: click the Tasks button and browse to Materials → Quantum Mechanics → Quantum ESPRESSO → Defect Formation Energy
To open this panel from the entry group for the results of a job .
.
The following licenses are required to use this panel: MS Maestro, Quantum Espresso Interface
- Using
- Features
- Additional Resources
Using the Defect Formation Energy Panel
This panel enables you to calculate the defect formation energy including a correction term for charged defects. The defect formation energy is calculated as follows:

 is the total energy of the optimized system containing the defect.
is the total energy of the optimized system containing the defect.  is the total energy of the optimized reference system (bulk or slab).
is the total energy of the optimized reference system (bulk or slab).  depends on the defect type, where
depends on the defect type, where  when the defect involves the addition of an atom to the system (e.g. an interstitial defect) and
when the defect involves the addition of an atom to the system (e.g. an interstitial defect) and  when the defect involves the removal of an atom from the system (e.g. a vacancy defect).
when the defect involves the removal of an atom from the system (e.g. a vacancy defect). is the defect chemical potential.
is the defect chemical potential.  is the total charge of the defect system.
is the total charge of the defect system.  is the Fermi energy, which, in this context, takes values between 0 and the band gap.
is the Fermi energy, which, in this context, takes values between 0 and the band gap.  is the valence band maximum energy.
is the valence band maximum energy.  is the defect correction energy for charged point defects calculated using the the Freysoldt, Neugebauer, Van de Walle (FNV) correction method as implemented in the sxdefectalign code [85].
is the defect correction energy for charged point defects calculated using the the Freysoldt, Neugebauer, Van de Walle (FNV) correction method as implemented in the sxdefectalign code [85].
The inputs required for this panel are electrostatic potential surface files (-.cub) for a defect structure and its corresponding bulk crystal. These can be generated efficiently using the Defect Setup Calculations Panel or the Quantum ESPRESSO Calculations Panel.
Point defects as, for instance, vacancies, substitutions, and interstitials, are ubiquitous in any crystal whether unintentional or by design (e.g. doping). Even at small concentrations they strongly affect electronic, structural and mechanical properties of crystals. Periodic density functional theory (DFT) provides a valuable insight into point defects structures and their energetics. However, special care is needed in studies of locally charged defects whose energetic properties slowly converge with the model cell size. The artificial long-range Coulomb interactions between charged defects must be corrected for accurate and converged defect formation energies. The FNV correction became particularly prominent as it only requires two supercell calculations and it avoids non-interpretable fitting constants [86]. This panel allows us to calculate the full correction term,  , and adds it to the defect formation energy of the charged defect. A difference between the bulk and defect structures potential surfaces is also provided and can be visualized in the Workspace.
, and adds it to the defect formation energy of the charged defect. A difference between the bulk and defect structures potential surfaces is also provided and can be visualized in the Workspace.
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
Defect Formation Energy Panel Features
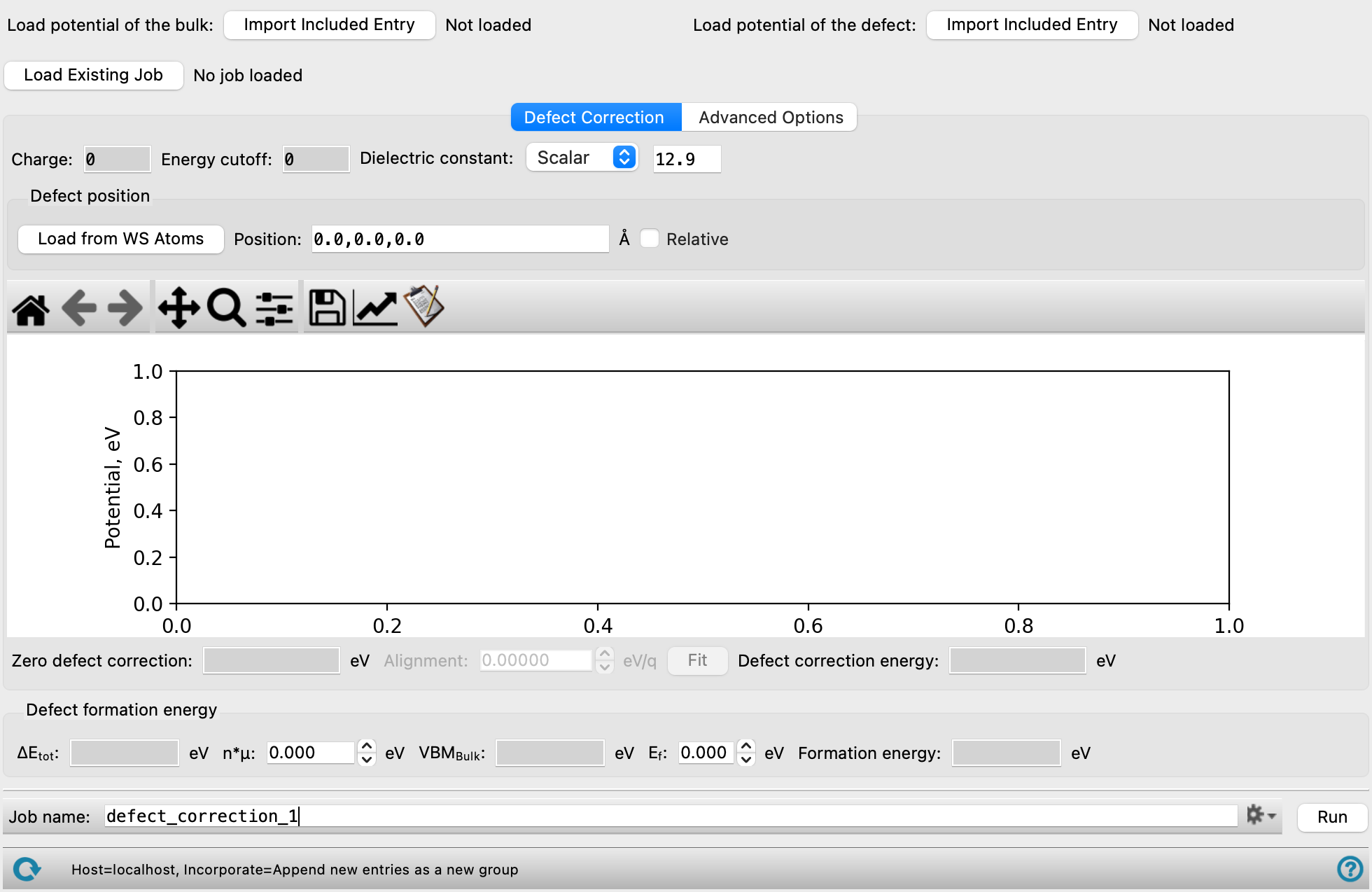
- Load potential of the bulk button
-
Click the Import button to load a bulk crystal structure from the Workspace with an associated potential surface (
-.cub) calculated from either the Defect Setup Calculations Panel or Quantum ESPRESSO Calculations Panel. - Load potential of the defect button
-
Click the Import button to load a crystal structure with a defect from the Workspace with an associated potential surface calculated from either the Defect Setup Calculations Panel or Quantum ESPRESSO Calculations Panel.
- Load Existing Job button
-
Load a defect correction output file generated using the Defect Correction panel. The file can be found in the job directory with the extension
-.spxgz. Click to open the Load Existing Job dialog box, where you can navigate to the file. The name of the file you selected is displayed to the right of the button. - Defect Correction tab
-
Plot the defect-induced potential calculated by DFT, the long-range potential based on a model charge density, as well as the short-range potential obtained from the difference of the DFT and the long-range part to understand the defect correction energy. The plot can be loaded from an existing job or generated by running a calculation using this panel.
- Charge text box
-
Displays the charge of the system specified in the Defect Setup Calculations Panel. Noneditable.
- Energy cutoff text box
-
Displays the reciprocal space cutoff energy specified in the Defect Setup Calculations Panel. Noneditable.
- Dielectric constant option menu and text box
-
Select a dielectric constant option to scale the energies and potentials by. The options include:
-
Scalar—For isotropic screening, select this option and enter the dielectric constant.
-
Tensor—For anisotropic screening, select this option and enter the dielectric tensor.
If the atoms in the cell are allowed to relax after introducing the defect, which is true when using the Defect Setup Calculations Panel, the electronic and the ionic contribution to the dielectric constant are needed. If the atoms in the cell are not allowed to relax during the calculation, only the electronic contribution to the screening of the defect charge needs to be considered.
-
- Defect position section
-
- Load from WS Atoms button
-
Specify the atoms that define the defect in the Workspace and click this button to locate it.
- Position text box
-
Display the position of the defect in the cell in angstroms. The position is automatically populated from the imported potential file for the defect and is noneditable if it was created using the Defect Setup Calculations Panel.
- Relative option
-
Check this option to use relative coordinates for the defect position. Otherwise, angstroms are used.
- Plot toolbar
-
The toolbar has tools for manipulating the plot and for saving images. The buttons that are common to all plot toolbars are described in the Plot Toolbar topic.
- Plot area
-
This area displays the plot of the potential (eV) against distance from the center of the defect (angstrom). The defect-induced potential calculated by DFT, the long-range potential based on a model charge density, as well as the short-range potential obtained from the difference of the DFT and the long-range part are plotted. For more details see [85] and [86]. Under the assumption that the short-range potential decays to zero between the original defect and its periodic images, the alignment term can be extracted by the plateau of the "defect-bulk-long range" part. You can adjust the range of distances used for calculating the alignment.
- Zero defect correction text box
-
Displays the contribution of the macroscopically screened lattice energy of the defect charge with compensating background, in electron volts. Noneditable.
- Alignment text box and Fit button
-
The alignment term is calculated from fitting the long range bulk defect potential in the plot, in eV/q. You can adjust the range of distances used for calculating this value, by dragging the solid vertical lines on the plot. Click Fit to update the value based on the selected fitting region.
- Defect correction energy text box
-
Displays the defect correction energy calculated from the zero defect energy and alignment term, in electron volts, as follows: Zero defect correction + (Charge * Alignment). Noneditable.
- Advanced Options tab
-
Specify additional parameters for the calculation. These parameters typically help smooth noisy plots.
- Average text box
-
Specify a value, in angstrom, to use for averaging in order to decrease oscillations in plots.
- Atom average text box
-
Specify a value, in angstrom, for Gaussian broadening for atomic-sphere averages
- Anticore option
-
Smooth the electrostatic potential by removing ionic cores from the potential. This increases the length of the calculation.
- Defect formation energy section
-
View and specify parameters for the defect formation energy calculation. For more information on the parameters of the calculation, see the Using section of the documentation.
- ΔEtot text box
-
Displays the difference in total energy between the optimized defect and bulk structures, in eV. Noneditable.
- n*μ text box
-
Specify the value of n*μ to use in calculating the formation energy, in eV. ni depends on the defect type, where ni = 1 when the defect involves the addition of an atom to the system (e.g. an interstitial defect) and ni = -1 when the defect involves the removal of an atom from the system (e.g. a vacancy defect). μi is the defect chemical potential which can be determined from experiment or other calculations. The specifics of the experimental conditions are incorporated into the defect energy calculations through the chemical potential value. Though the choice of chemical potential strongly influences the absolute defect formation energy, it does not affect the relative energies of the defects' different charged states.
- VBMBulk text box
-
Displays the valence band maximum as calculated for the bulk system, in eV. Noneditable.
- Ef text box
-
Specify the Fermi energy between 0 and the band gap, in eV.
- Formation energy text box
-
Displays the defect formation energy in eV. Noneditable.
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Defect Formation Energy - Job Settings Dialog Box, where you can make settings for running the job.
- Status bar
-
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel.If you can submit a job from the panel, the status bar displays information about the current job settings and status for the panel. The settings include the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
The status bar also contains the Help button
 , which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.
, which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.