Optoelectronic Film Properties Panel
dipole moment, order parameter, singlet exciton energy transfer, seet, intersystem crossing, isc, refractive index, extinction coefficient, ellipsometry
Calculate various optoelectronic film properties, such as transition dipole moment order parameter, singlet exciton energy transfer rate, intersystem crossing rate, refractive index, and extinction coefficient from molecular excited state properties.
To open this panel: click the Tasks button and browse to Materials → Quantum Mechanics → Optoelectronic Film Properties → Optoelectronic Film Properties
The following licenses are required to use this panel: MS Maestro, Jaguar
- Using
- Features
- Additional Resources
Using the Optoelectronic Film Properties Panel
This panel allows you to determine various optoelectronic film properties such as order parameter, singlet exciton energy transfer rate, singlet-triplet intersystem crossing rate, refractive index, and extinction coefficient. Learn more about how each of these are calculated:
Transition Dipole Moment Order Parameter Tab
Order parameter is calculated using the transition dipole moment vector as the descriptor. The transition dipole moment can be calculated from TDDFT calculations of excitations, or manually defined.
The order parameter is calculated as (1/N) Σi=1,N P2(cosθi), where cos θi is determined from the scalar product of a vector for the descriptor and the vector for the director, and P2(cosθi) is the second-order Legendre polynomial, (3 cos2θi−1)/2. The sum is taken over all descriptor vectors defined for all molecules. Thus, the order parameter is the average value of the Legendre polynomial over all descriptors. The use of P2 means that the order parameter does not distinguish between descriptor vectors that are in opposite directions. The significance of the order parameter for some critical values is given below:
- 1 : perfect alignment along the director, either parallel or antiparallel
- −1/2 : perfect alignment perpendicular to the director.
- 0 : random orientation with respect to the director; or isotropic orientation; also perfect alignment at the "magic angle" (54.7°) where P2 is zero.
A zero value—or any value between the limits of 1 and −1/2—can be arrived at in multiple ways. For example, a random or isotropic set of orientations would have a zero order parameter. A set in which 1/3 of the descriptors were aligned along the director and 2/3 were aligned perpendicular to the director would also give a zero order parameter. If one is looking for small deviations from perfect alignment, values in the middle of the range indicate that the desired alignment was not found, regardless of the actual distribution of alignments.
The order parameter is returned as a Material Science property, TDM X Order Parameter, where X indicates whether the state is a singlet or triplet (S or T) and the excited state level (1, 2, 3...). If the Choose two atoms to define the transition moment option is selected, X is 1. The property is added to the product structures.
Singlet Exciton Energy Transfer Rate Tab
The singlet exciton energy transfer rates ( ) are calculated for dimers in an organic film. The dimers are identified as all the donor-acceptor molecule pairs in the input cell. The rates are calculated using the semiclassical Marcus equation:
) are calculated for dimers in an organic film. The dimers are identified as all the donor-acceptor molecule pairs in the input cell. The rates are calculated using the semiclassical Marcus equation:

where  is the electronic coupling between the donor and acceptor molecules and is estimated using the Förster dipole-dipole approximation,
is the electronic coupling between the donor and acceptor molecules and is estimated using the Förster dipole-dipole approximation,  is the change in energy, and
is the change in energy, and  is the reorganization energy. The reorganization energy can be obtained from DFT calculations or literature values. To calculate the reorganization energy, the energy difference between S0 of the S0 and S1 geometry optimized donor molecule must be added to the energy difference between S1 of the S0 and S1 geometry optimized acceptor molecule. This can be done using the QM Multistage Workflow Panel or the Optoelectronics Calculations Panel.
is the reorganization energy. The reorganization energy can be obtained from DFT calculations or literature values. To calculate the reorganization energy, the energy difference between S0 of the S0 and S1 geometry optimized donor molecule must be added to the energy difference between S1 of the S0 and S1 geometry optimized acceptor molecule. This can be done using the QM Multistage Workflow Panel or the Optoelectronics Calculations Panel.
The singlet exciton energy transfer rate can then be analyzed as a function of intermolecular distance.
Note that singlet exciton energy transfer rates are impacted by intermolecular orientations and hence by film morphology. For improved agreement with experimental measurements, it is strongly suggested to match the model organic film morphology to the experimental conditions used in the lab (e.g., vacuum deposition or solution processing).
Singlet-Triplet Intersystem Crossing Rate Tab
The singlet-triplet intersystem crossing (ISC) rate and reverse intersystem crossing (RISC) rate are calculated for the selected molecule type. In this panel, the rates are calculated using both the semiclassical Marcus equation and semiclassical Marcus-Levich-Jortner equation.
Marcus theory, originally developed for electron transfer, has been adapted to describe non-radiative transitions such as ISC and RISC in organic light-emitting diodes (OLEDs). This theory treats these transitions as charge-transfer-like processes, where the rate depends on the reorganization energy (λ), the driving force (ΔG), as well as the coupling between the initial and final states. Marcus theory provides a framework for calculating ISC and RISC rates by linking them to the energy difference between singlet and triplet states (ΔEST), spin-orbit coupling, and reorganization effects.
The semiclassical Marcus-Levich-Jortner model extends Marcus theory by incorporating quantum mechanical effects of molecular vibrations. This model is particularly useful for systems where high-frequency vibrations (e.g., C–H and C–C stretching modes) significantly influence the transition rates. By accounting for both classical and quantum vibrational modes, the Marcus-Levich-Jortner model offers a more detailed description of ISC and RISC, capturing the subtleties of molecular interactions that impact OLED efficiency.
The reorganization energy of ISC from S1 to T1 is calculated by taking the energy difference between the triplet state at the singlet-optimized geometry and the triplet state at its own optimized geometry: λ=E(T1 at S1)−E(T1). The reorganization energy of RISC from T1 to S1 is calculated by taking the energy difference between the singlet state at the triplet-optimized geometry and the singlet state at its own optimized geometry: λ=E(S1 at T1)−E(S1). This can be done automatically in this workflow or manually using the QM Multistage Workflow Panel. It is recommended to calculate the reorganization energies using this panel but note that this increases the calculation time.
Refractive Index Tab
The frequency-dependent refractive index can be used to study birefringence in materials composed of molecules, such as liquid crystals and crystalline materials with anisotropic optical behavior. The Lorentz-Lorentz equation relates the refractive index and the polarizability of an isotropic material. The Vuks equation derives from the Lorentz-Lorentz equation under the assumption that the electric field experienced by a molecule in a material, which is in the presence of an electric field, is the same in all directions. As a result, anisotropies in the refractive index can be investigated as a function of the frequency of light of ordinary and extraordinary rays.
Extinction Coefficient Tab
The extinction coefficient, together with the refractive index, allows one to perform calculations in an area known as spectroscopic ellipsometry. Molar extinction coefficients of components in a material, and how they relate to the total absorption spectrum according to the Beer-Lambert law, can be calculated using this option.Birefringence in materials that may exhibit optical anisotropies, such as liquid crystals, can be investigated by breaking down the extinction coefficient for a component along user defined axes. This option also similarly provides the total absorption spectrum, as well as the breakdown, of the material as a whole. For finer detail, a histogram of excited state properties, like energies, transition dipole moments, oscillator strengths, etc., of individual molecules for a given component is provided.
To visualize the results, you can use the Optoelectronic Film Properties Viewer Panel (Click the Tasks button and browse to Materials → Quantum Mechanics → Optoelectronic Film Properties → Optoelectronic Film Properties Viewer).
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
Optoelectronic Film Properties Panel Features
- Use structures from option menu
- Open Project Table button
- File name text box and Browse button
- Transition dipole moment order parameter option
- Singlet exciton energy transfer rate option
- Singlet-triplet intersystem crossing rate option
- Refractive index option
- Extinction coefficient option
- Temperature text box
- Options button
- Transition Dipole Moment Order Parameter tab
- Singlet Exciton Energy Transfer Rate tab
- Singlet-Triplet Intersystem Crossing Rate tab
- Refractive Index tab
- Extinction Coefficient tab
- Job toolbar
- Status bar
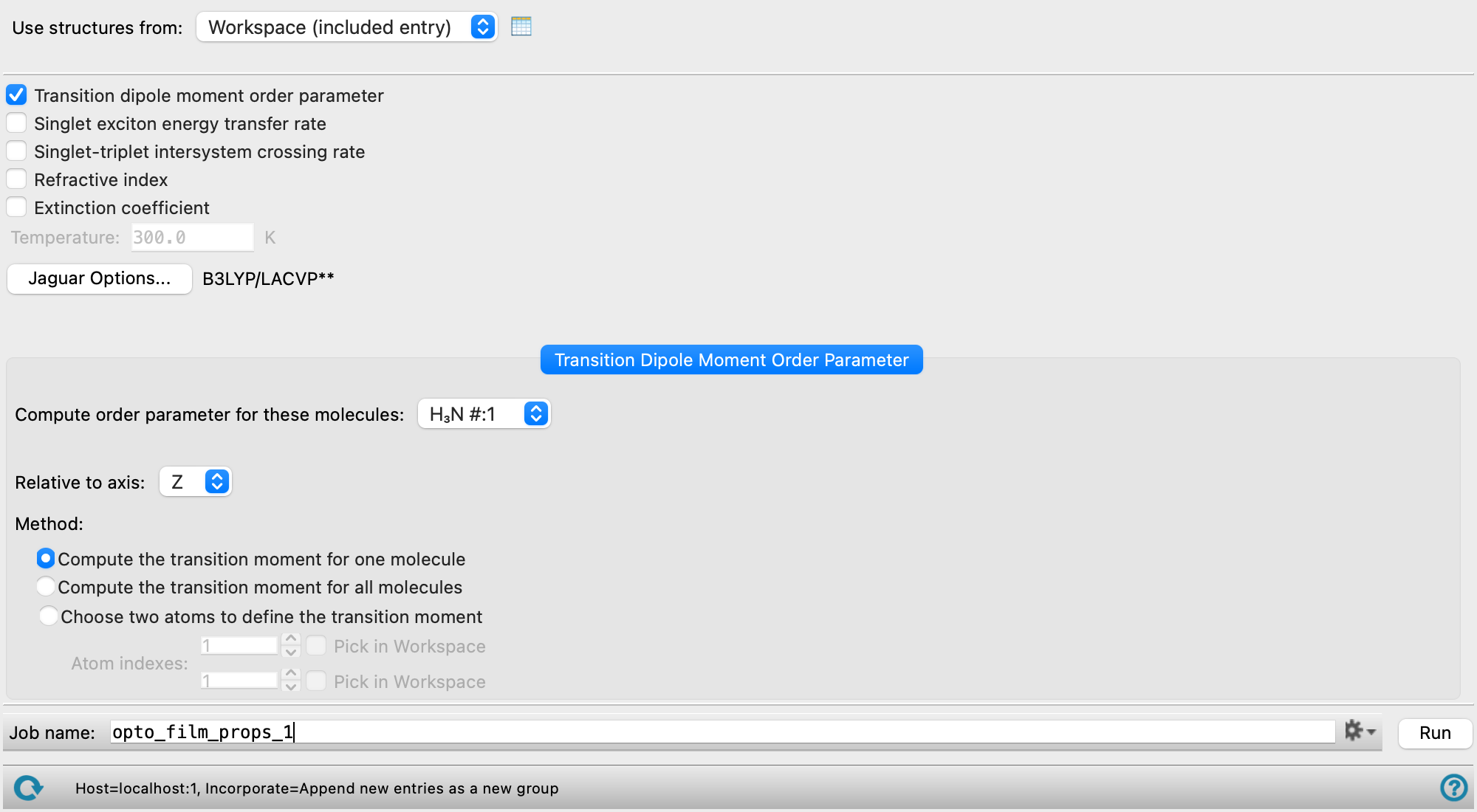
- Use structures from option menu
-
Choose the structure source for the order parameter calculation.
- Workspace (included entries)—Use the entries that are currently included in the Workspace, treated as separate structures.
- File—Use the specified file. When this option is selected, the File name text box and Browse button are displayed.
- Open Project Table button
-
Open the Project Table panel, so you can

- File name text box and Browse button
-
Enter the file name in this text box, or click Browse and navigate to the file. The name of the file you selected is displayed in the text box.
- Transition dipole moment order parameter option
-
Select this option to calculate the transition dipole moment order parameter. The Transition Dipole Moment Order Parameter tab is present when this option is selected.
- Singlet exciton energy transfer rate option
-
Select this option to calculate the singlet exciton energy transfer rate using the Förster dipole-dipole approximation. The Singlet Exciton Energy Transfer Rate tab is present when this option is selected.
- Singlet-triplet intersystem crossing rate option
-
Select this option to calculate the singlet-triplet intersystem and reverse intersystem crossing (ISC/RISC) rates using Marcus and Marcus-Levich-Jortner models. The Singlet-Triplet Intersystem Crossing Rate tab is present when this option is selected.
- Refractive index option
-
Select this option to calculate the frequency-dependent refractive index using the Vuks model. The Refractive Index tab is present when this option is selected.
- Extinction coefficient option
-
Select this option to calculate the extinction coefficient. The Extinction Coefficient tab is present when this option is selected.
- Temperature text box
-
Specify the temperature to be used, in kelvin. Only available when the Singlet exciton energy transfer rate option or Singlet-triplet intersystem crossing rate option is selected.
- Options button
-
Set Jaguar options for the TDDFT calculations. Opens the Jaguar Options - Transition Moment Order Parameter Dialog Box. This dialog box also allows you to specify additional Jaguar keywords. The solvent (if any), level of theory, and basis set are shown to the right of the button.
-
By default, 3 excited states are calculated (nroot=3).
- Transition Dipole Moment Order Parameter tab
-
Specify the molecule for which and the method by which the order parameter is calculated. Only present when the Transition dipole moment order parameter option is selected.
- Compute order parameter for these molecules option menu
-
Select the molecule type for which the order parameter should be computed. The Find Molecule Types button is present for structures with greater than 20000 atoms. Click the button to calculate the molecule types to choose from for the calculation.
- Relative to axis option menu
-
Select the axis to use as the vector for the director, from X, Y , or Z. The angle of the transition dipole moment vector relative to the selected axis is used in the order parameter calculation.
- Method options
-
Select how the transition dipole moment vectors used in the order parameter calculations should be computed.
- Compute the transition moment for one molecule option
-
A TDDFT excited state calculation is performed on one randomly selected molecule from the Compute order parameter for these molecules option menu. Parameters for the TDDFT calculation can be set through the Jaguar Options button. The transition dipole moment is taken from the lowest singlet excited state, and projected on all the other instances of that molecule in the system.
- Compute the transition moment for all molecules option
-
TDDFT excited state calculations are performed for all molecules selected from the Compute order parameter for these molecules option menu. Parameters for the TDDFT calculation can be set through the Jaguar Options button. The transition dipole moment is taken from the lowest singlet excited state.
This is the most computationally intensive option if there is more than one instance of the molecule, as a separate TDDFT calculation is launched for each instance.
- Choose two atoms to define the transition moment option
-
Manually define the transition dipole moment vector of a molecule as the vector between two atoms. This definition is used for each instance of the molecule. You should only select this method if you have prior knowledge of the expected transition dipole moment.
A TDDFT calculation is not performed if this option is selected and the Jaguar Options button is not available.
- Atom indexes text boxes and Pick in Workspace options
-
Select the two atoms which define the transition dipole moment vector in the Workspace. You can manually enter the atom indices in the text boxes. One index should be entered in each text box. You can also select the Pick in Workspace option to pick the atom in the Workspace. Select the Pick in Workspace option to the right of the first text box to pick the index for the first atom, and then select the Pick in Workspace option to the right of the second text box to pic the index for the second atom. The index should automatically update in the text box.
- Singlet Exciton Energy Transfer Rate tab
-
Specify the dimer and corresponding parameters for the singlet exciton energy transfer rate calculation. Only present when the Singlet exciton energy transfer rate option is selected.
- Donor molecules option menu
-
Select the molecule type to be used as a donor in the energy transfer. The Find Molecule Types button is present for structures with greater than 20000 atoms. Click the button to calculate the molecule types to choose from for the calculation.
- Acceptor molecules option menu
-
Select the molecule type to be used as an acceptor in the energy transfer. The Find Molecule Types button is present for structures with greater than 20000 atoms. Click the button to calculate the molecule types to choose from for the calculation.
- Donor singlet excitation energy correction text box
-
Specify the correction to the donor S1 excitation energy in electron volts. The default value is 0 eV.
-
The correction value can be obtained from high-level calculations or experimental values which allows use of an inexpensive level of theory with high accuracy. This also allows for relaxation effects in the excited state (e.g. S1*-->S1) to be accounted for.
- Acceptor singlet excitation energy correction text box
-
Specify the correction to the acceptor S1 excitation energy in electronvolts. The default value is 0 eV.
-
The correction value can be obtained from high-level calculations or experimental values which allows use of an inexpensive level of theory with high accuracy. This also allows for relaxation effects in the excited state (e.g. S1*-->S1) to be accounted for.
- Reorganization energy text box
-
Specify the S1 reorganization energy. The default value is 0.1 eV.
-
The reorganization energy can be obtained from DFT calculations or literature values, see the Using section for more information.
- Singlet-Triplet Intersystem Crossing Rate tab
-
Specify the molecule type and corresponding parameters for the singlet-triplet intersystem crossing rate calculation. Only present when the Singlet-triplet intersystem crossing rate option is selected.
- Compute rate for these molecules option menu
-
Select the molecule type for which the intersystem crossing rate should be computed. The Find Molecule Types button is present for structures with greater than 20000 atoms. Click the button to calculate the molecule types to choose from for the calculation.
- Estimate crossing reorganization energies option and section
-
Select this option to specify the crossing reorganization energies rather than calculate them as part of the workflow. See the Using section for more information.
- Intersystem crossing reorganization energy text box
-
Specify the reorganization energy of the triplet with respect to the singlet used to calculate the ISC rate in electron volts.
- Reverse intersystem crossing reorganization energy text box
-
Specify the reorganization energy of the singlet with respect to the triplet used to calculate the RISC rate in electron volts.
- Quantum mechanical reorganization energy text box
-
Specify the quantum-mechanical contribution to the reorganization energy, which is the portion of the total reorganization energy associated with high-frequncy vibrational modes, which is used to calculate the Huang-Rhys parameters.
- Refractive Index tab
-
Specify the parameters for the refractive index calculation. Only present when the Refractive index option is selected.
- Compute the polarizability for a single molecule of each type option
-
Calculate the polarizability tensor for a single representative molecule of each type. Rotate the representative polarizability tensor onto the frame of reference for all other molecules of the same type.
- Compute the polarizability for all molecules of each type option
-
Calculate the polarizability tensor directly for each molecule in the system. This option can increase calculation time significantly.
- Starting frequency text box
-
Specify the starting frequency in eV for frequency-dependent polarizability calculations.
- Step size text box
-
Specify the step size in frequency in eV for frequency-dependent polarizability calculations.
- Number of frequencies text box
-
Specify the number of frequencies to calculate. This value along with the Starting frequency and Step size defines the end point of the range of frequencies used in the polarizability calculations.
- Mass density text box
-
Specify the mass density of the material in g/cm3.
- Refractive index axes option menu
-
Specify the vectors (i, j, and k) along which the refractive index is calculated.
- xyz selected—calculates components along the x, y, and z directions.
- cell selected—calculates components along the a, b, and c lattice vectors of the input cell. If the cell is orthorhombic and a, b, and c are along x, y, and z then this option is equivalent to using the xyz option.
- diag selected—diagonalizes the polarizability tensor at each frequency and chooses the vectors from that with the largest eigenvalue.
- Extinction Coefficient tab
-
Specify the parameters for the extinction coefficient calculation. Only present when the Extinction coefficient option is selected.
- Compute the transition dipole moment for a single molecule of each type option
-
Calculate the transition dipole moment for a single representative molecule of each type. Rotate the representative dipole moment onto the frame of reference for all other molecules of the same type.
- Compute the transition dipole moment for all molecules of each type option
-
Calculate the transition dipole moment directly for each molecule in the system. This option can increase calculation time significantly.
- Extinction coefficient axes option menu
-
Specify the vectors (i, j, and k) along which the extinction coefficient is calculated.
- xyz selected—calculates components along the x, y, and z directions.
- cell selected—calculates components along the a, b, and c lattice vectors of the input cell. If the cell is orthorhombic and a, b, and c are along x, y, and z then this option is equivalent to using the xyz option.
- Frequency bin width text box
-
Specify the bin value for the excited state energies of molecules to plot the absorption spectrum.
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Optoelectronic Film Properties - Job Settings Dialog Box, where you can make settings for running the job.
- Status bar
-
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel.If you can submit a job from the panel, the status bar displays information about the current job settings and status for the panel. The settings include the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
The status bar also contains the Help button
 , which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.
, which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.