Reaction Network Enumeration Profiler Panel
Set up and run a set of optimizations with Jaguar on structures that are reactants, transition states, intermediates, or products in a reaction network enumeration, with an optional conformational search
To open this panel: click the Tasks button and browse to Materials → Quantum Mechanics → Reaction Network → Reaction Network Enumeration Profiler Calculations
The following licenses are required to use this panel: MS Maestro, MS Reactivity, OPLS (optional), Jaguar, MS Force Field Applications (optional)
- Using
- Features
- Additional Resources
Using the Reaction Network Enumeration Profiler Panel
The Reaction Network Enumeration Profiler (RxnEnumProfiler) Panel allows users to simultaneously create and run multiple reaction networks. Instead of submitting multiple jobs from the Reaction Network Profiler Panel, you can set up these jobs from the RxnEnumProfiler panel. This enables you to study a library of catalyst candidates or reactions by leveraging R-group enumeration and swap features (enabling complete swapping of catalysts - be it the metal center itself or entire moieties). You can also generate cheminformatics descriptors that can be used in a machine learning (ML) model.
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
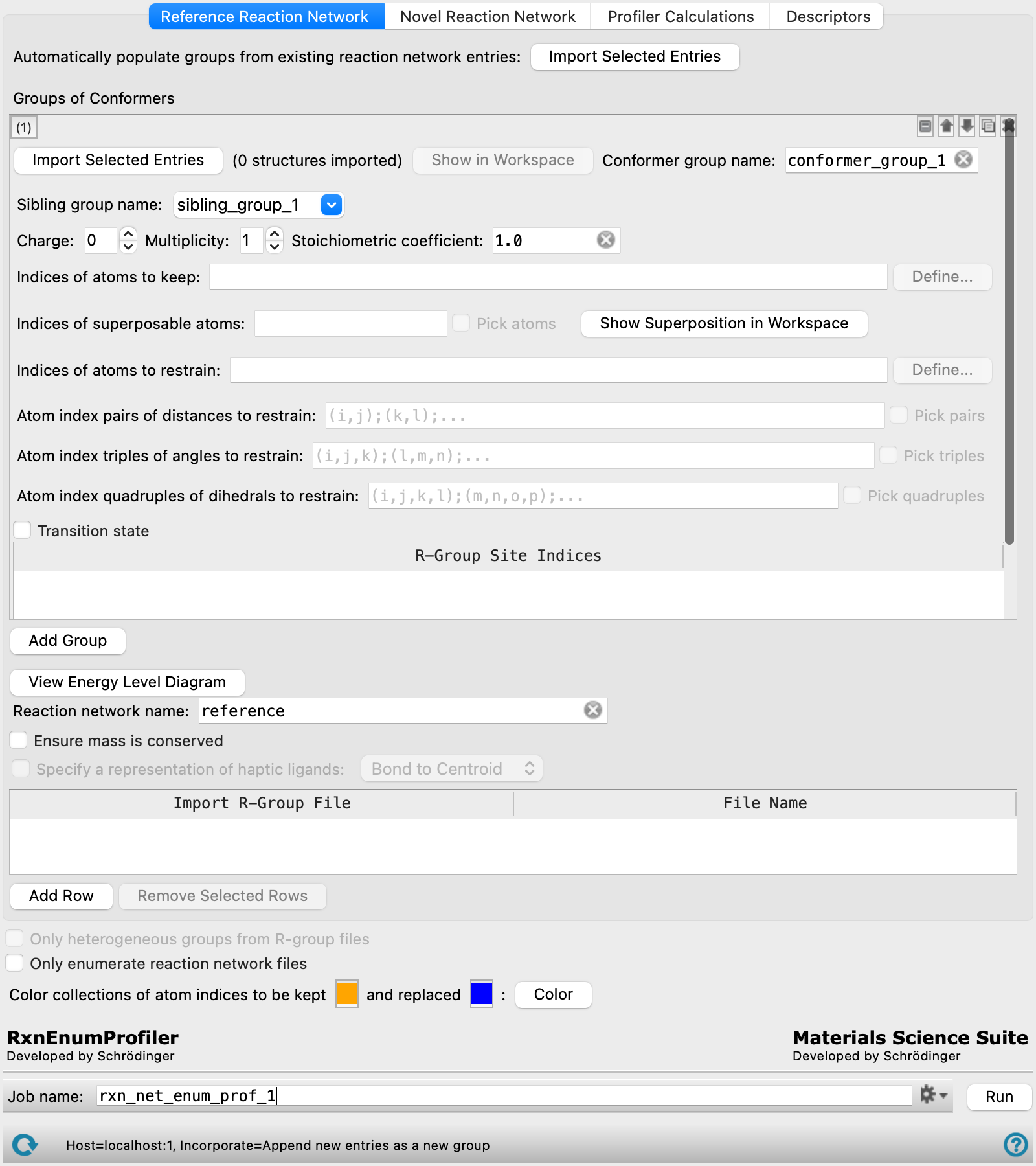
Reference Reaction Network Tab
This tab sets up structures for the reaction network, in which the structures are optimized to locate a minimum or a transition state. The main purpose of the tab is to specify restraints on atoms or geometric parameters in the process of finding the minimum or the transition state. These restraints (and other settings) are stored as entry properties in the output structures.
The tab operates on groups of conformers. Each group must have the same connectivity and atom numbering, but you can include stereoisomers (R,S or E,Z) as well as different geometries of rotatable groups. The group can of course consist of a single structure. Settings are made for each group of conformers, and the groups are stored in the Project Table.
The controls for these settings can be added for a new group (click Add Group), or duplicated from an existing group (click the Duplicate button  ). The order of the groups is irrelevant to the calculations done in the reaction network: each group is processed independently. You can therefore rearrange the groups as you like, using the tools at the top right of the group controls.
). The order of the groups is irrelevant to the calculations done in the reaction network: each group is processed independently. You can therefore rearrange the groups as you like, using the tools at the top right of the group controls.
A stage in the reaction network can consist of multiple conformer groups. The conformer groups in a stage are "siblings": they have the same parent stages (and also the same child stages). This allows you to carry a structure through several reaction stages where it is a spectator until it becomes a reactant in a particular stage, and likewise to carry a product or byproduct through subsequent stages of the reaction. Introducing the spectators allows you to define stages that have the same stoichiometry, which in turn allows you to compare energies at each stage to obtain a reaction profile.
For transition states, a preoptimization is performed with Jaguar prior to doing the transition state search, using specified distance, angle, and dihedral restraints. In the transition state search, these restrained coordinates become active coordinates that are used to guide the transition state search, which is performed without restraints. In this way, the non-active coordinates can be optimized before the transition state is performed.
For reactants, products, and intermediates, where the optimization is to a minimum, it is usually not a good idea to apply restraints, as only a single optimization step is performed.
Novel Reaction Network Tab
Given a reaction scheme defined in the Reference Reaction Network tab, this tab allows you to make substitutions on the substrate or the catalyst in the stages of this scheme. The structures for each step of the input reaction scheme are treated as a set of reference structures, and the new catalyst or substrate is treated as the novel structure. For the novel structure and each reference structure, you designate atoms to keep (or remove) in the new structure, then specify how the structures are to be aligned so that the correct bonds are formed and the fragments have the desired relative orientation.
Reaction Network Tab
This tab is used to run Jaguar calculations on structures that form parts of a reaction network, which have been defined in the preceding tabs. There is no restriction on the number of structures or the type of structures, so you could process several reactions at the same time. However, the output is returned in a single entry group named with the job name. This entry group contains entry groups of the optimized structures that match the entry groups of the structures that are selected as input. You can view a reaction profile in the Reaction Profile Viewer Panel.
If subjobs fail for some structures, you can import these structures from the jobname_failed.maegz file. Each structure has a set of Successful step-name Boolean properties, which you can use to determine which steps in the workflow failed. As these are secondary properties you will have to show them in the Project Table first—see Organizing Properties for more information.
The structure output file is named jobname-out_rxnwf.mae, and contains the geometries of the conformers generated at the critical points. The geometries are the optimized or transition state geometries. It also contains energy properties from all of the given stages, including analysis stages. These properties have a stageN suffix, to identify the stage in the network.
In addition to the structure output, there are other output files, jobname_conf_avg_rel_reactants.csv and jobname_conf_avg_rel_parents.json, that contain conformationally averaged energies relative to reactants and relative to parents for points along the reaction path. If multiple temperatures and pressures are specified, the reaction rate for each temperature and pressure is written to a CSV file, jobname_conf_avg_rate_constants.csv.
The job calculates turn over frequencies using the energetic span model by default. The output file jobname_tof.csv reports both the sum-over-states turn over frequencies and the determining states approximated turn over frequencies, along with information on the energy type, whether conformationally averaged energies or the lowest conformer energy were used, reaction energy, and the determining intermediate, TS, and energy span.
Reaction Network Enumeration Profiler Panel Features
- Reaction Network Enumeration Profiler Panel — Reference Reaction Network Tab
- Reaction Network Enumeration Profiler Panel — Novel Reaction Network Tab
- Reaction Network Enumeration Profiler Panel — Profiler Calculations Tab
- Reaction Network Enumeration Profiler Panel — Descriptors Tab
- Job toolbar
- Status bar
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Reaction Network Enumeration Profiler- Job Settings Dialog Box, where you can make settings for running the job.
- Status bar
-
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel.If you can submit a job from the panel, the status bar displays information about the current job settings and status for the panel. The settings include the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
The status bar also contains the Help button
 , which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.
, which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.