Compute KMC Hopping Parameters Panel
Compute charge hopping parameters in an amorphous solid or a crystalline solid using Jaguar and QSite calculations.
To display this panel
The following licenses are required to use this panel: MS Maestro, MS Mobility, Jaguar, QSite
- Using
- Features
- Additional Resources
Using the Compute KMC Hopping Parameters Panel
The charge hopping parameters are computed in several stages. First, data on the charged and uncharged forms of each molecule in the cell is generated using QSite (see the QSite User Manual — Contents). The calculations are done with one molecule in the QM region and its neighbors in the MM region. These calculations provide the site energies, which are the differences in energy between the charged and uncharged form. Next, the coupling integral is evaluated between each molecule and its neighbors, using Jaguar. The techniques are the same as used in the Electron Coupling Panel.The QSite wave functions are used to evaluate the coupling, as these contain the effects of the neighbors.
The calculation of the hopping parameters involves a large number of QM calculations: four QSite calculations for each molecule, and two Jaguar calculations for each pair of neighboring molecules (for hopping in each direction). These calculations can be distributed across multiple processors, as the QSite calculations are independent of each other, and likewise for the Jaguar calculations. In order to reduce the turnaround time, it is recommended that you run on a host with many nodes, so you can distribute the load as much as possible.
It is possible that some of the QSite calculations may fail to converge. The results of the failed jobs are returned to the host on which you started the job, and you can use the Restart Hopping Parameter Calculations Panel to restart these jobs with keyword settings to improve convergence.
If subjobs fail for some structures, you can import these structures from the jobname_failed.maegz file. Each structure has a set of Successful step-name Boolean properties, which you can use to determine which steps in the workflow failed. As these are secondary properties you will have to show them in the Project Table first—see Organizing Properties for more information.
The hopping parameter data from the Jaguar and QSite calculations is stored in SQL databases, one for each field strength and charge type. The databases also contain information on the structure used for the calculations.
Compute KMC Hopping Parameters Panel Features
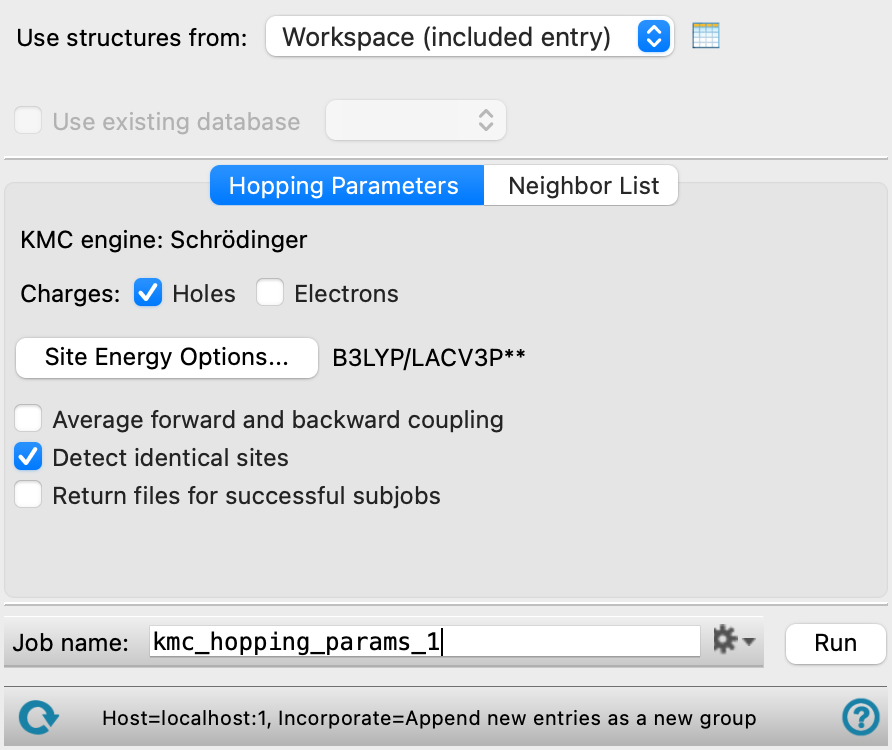
- Use structure from option menu
-
Choose the structure source for computing hopping parameters. If the structure source contains multiple structures, only the first structure is used. The structure must have periodic boundary condition properties.
- Workspace (included entry)—Use the entry that is currently included in the Workspace. Only one entry must be included in the Workspace.
- File—Use the specified file. When this option is selected, the File name text box and Browse button are displayed.
- Database—Use the structure whose identity is recorded in the specified SQL database. When this option is selected, the File name text box and Browse button are displayed, so you can specify the database.
- Open Project Table button
-
Open the Project Table panel, so you can

- File name text box and Browse button
-
Enter the file name in this text box, or click Browse and navigate to the file. The name of the file you selected is displayed in the text box.
- Use existing database option and menu
-
If the structure in the Workspace has any hopping parameter databases associated with it, use the database chosen from the menu, and do not calculate the charge hopping parameters. This option is present if you choose Workspace (included entry) or File from the Use structures from option menu.
- Hopping Parameters tab
-
Set options for the charge hopping calculations.
- KMC engine text
-
Displays the KMC engine in use, Schrödinger.
- Charges options
-
Select the type of charge carrier (Holes and/ or Electrons) for the hopping parameters calculation.
- Missing data report
-
This text reports the number of sites that are missing and the number of pairs that are missing from the hopping parameter database. Only present when the Use existing database option is selected.
- Options button
-
Set Jaguar options for the site energy calculations. These settings are passed to QSite for the QM part of the calculation. Opens the Jaguar Options - Compute KMC Charge Mobility Dialog Box. This dialog box also allows you to specify additional Jaguar keywords. The solvent (if any), level of theory, and basis set are shown to the right of the button.
- Average forward and backward coupling option
-
Select this option to average the forward and backward coupling energies calculated.
- Detect identical sites option
-
Detect sites that are identical due to crystal symmetry. Select this option for charge hopping calculations in a crystal; deselect it for amorphous materials.
- Return files for successful subjobs option
-
Return the files generated by successful subjobs. The returned files are gzipped tar archives, one for each subjob. By default only the files for unsuccessful subjobs are returned, so you can analyze them to find out why they failed. The charge hopping calculations involve a large number of QSite and Jaguar subjobs, so the quantity of output can be very large, and by default it is not returned if the subjob is successful.
- Neighbor List tab
-
Set options for determining which molecules are neighbors.
- Cutoff distance text box and based on option menu
-
Specify the cutoff distance in angstroms, and choose whether the distance is calculated for heavy atoms (i.e. not hydrogen) or all atoms. Only available when the Use existing database option is selected.
- Reset button
-
Reset the cutoff distance and basis to the defaults. Only available when the Use existing database option is selected.
- Update Statistics button
-
Apply the cutoff to determine the neighbor list and generate statistics for the nearest neighbors. Only available when the Use existing database option is selected.
- Statistics report
-
Reports the average, maximum, and minimum numbers of nearest neighbors, and the total number of neighbor pairs.
- Highlight button
-
Highlight the molecule mentioned in the statistics in the Workspace. The molecule is represented as tubes, and colored red; its neighbors are colored yellow. You might want to color all the molecules with a color that does not obscure this highlighting.
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Compute KMC Hopping Parameters - Job Settings Dialog Box, where you can make settings for running the job.
- Status bar
-
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel.If you can submit a job from the panel, the status bar displays information about the current job settings and status for the panel. The settings include the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
The status bar also contains the Help button
 , which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.
, which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.