Electron Coupling Panel
charge transfer
Compute the charge transfer rate between molecules in a structure that consists of multiple molecules (monomers). The structure could be generated from a molecular dynamics simulation with Desmond, for example. Marcus theory is used for the hopping rate, based on DFT calculations for the electron transfer between donors and acceptors with either full wave function or dimer frontier orbital splitting calculations of the coupling matrix element.
To open this panel, click the Tasks button and browse to Quantum Mechanics → Electron Coupling.
The following licenses are required to use this panel: MS Maestro, Jaguar
- Using
- Features
- Additional Resources
Using the Electron Coupling Panel
This panel can be used to calculate carrier hopping rates in an amorphous structure. It is mainly designed for the results of an MD simulation with a homogeneous system, but it can be used for any structure consisting of multiple molecules. Marcus theory is used for the hopping rate, based on DFT calculations for the electron transfer between donors and acceptors with either full wave function or dimer frontier orbital splitting calculations of the coupling matrix element. Ref. [2] gives an excellent review of this area.
To set up the structure with MD, you can use the Disordered System Builder Panel and then run a simulation with the MD Multistage Workflow Panel. You can also import structures from other sources into Maestro. The structures must consist of multiple molecules.
As well as supplying the structures, you must also supply the reorganization energy. You can calculate the reorganization energy with the Optoelectronics Calculations Panel. The reorganization energy is assumed to have been calculated for the isolated, ground state relaxed molecule, and is assumed to be independent of the geometry in the mixture. If you have a heterogeneous system, you must decide how to treat the reorganization energy, which is specified here as a single value and thus cannot be given for different components. For charge separation, the reorganization energy is not calculated directly with the Optoelectronics Calculations Panel, but can be calculated from the results as the sum of the Hole Small Polaron Stabilization Energy and Electron Small Polaron Stabilization Energyproperties in the Project Table.
The matrix elements for the hopping are calculated using QM wave functions based on DFT calculations. The two alternatives are to run calculations on the donor and acceptor in a pair independently, with the charge localized on one or the other of the two, or to run calculations on the dimer and constrain the charge to lie on either the donor or acceptor. The former is done with normal DFT methods; the latter is done with constrained DFT (CDFT). All calculations are run as single-point Jaguar calculations at the geometry of the input structure.
There is a chance that a QM calculation can result in an excited state rather than the ground state. CDFT calculations appear to be particularly susceptible to this issue. Therefore, it is always important to check the HOMO and LUMO values listed in the Project Table for the initial and final states to verify that the HOMO energy is lower than the LUMO energy. If these values are reversed, the calculation should be repeated, adding the setting vshift=0.0 in the Keywords text box. By default, the value is 0.2 for hybrid DFT functionals and 0.3 for pure DFT.
When the job is run, each Jaguar job is run sequentially, but you can run individual Jaguar jobs in parallel. Doing this is useful if the molecule or the basis set is large. You cannot run CDFT (dimer) jobs in parallel, but you can run dimer splitting jobs or DFT (monomer) jobs in parallel. If the job ends prematurely due to problems like network or hardware issues, you can restart it with the Restart Workflow Panel.
The results of the calculation depend on the coupling method chosen. For each dimer, a set of structures is produced.
-
For the dimer frontier orbital splitting method, the structural output consists of a dimer structure. The structure includes properties for the hopping rate and the coupling matrix element (in eV), as well as the Jaguar properties for the neutral dimer.
-
For the full wave function treatment, the output includes two dimer structures in addition to the DFT or CDFT structures. The dimer structures have the rate properties, one for the forward rate and one for the reverse rate. The properties are the charge transfer coupling (T_i->f in the Jaguar output) and the hopping rate. If you choose to calculate both hole and electron rates, there is one set of structures for each carrier type.
The hopping rate is defined by
K = (2π/ℏ) exp[−(L+ΔE)2/(4πLkT)] (Hab)2/√(4πLkT)
where L is the reorganization energy, ΔE is the charge hopping reaction energy, Hab is the electron transfer coupling matrix element, k is the Boltzmann constant and T is the temperature, and all energies are given in joules. The rate is in units of s−1. The Charge Hopping Rate computed by the Charge separation method represents the rate of spontaneous charge generation when two neutral molecules transform into a hole/electron pair, and is typically zero due to the large positive energy change (ΔE) for this process.
Once you have the results, you can use them in the Charge Mobility Panel to calculate the charge mobility.
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
Electron Coupling Panel Features
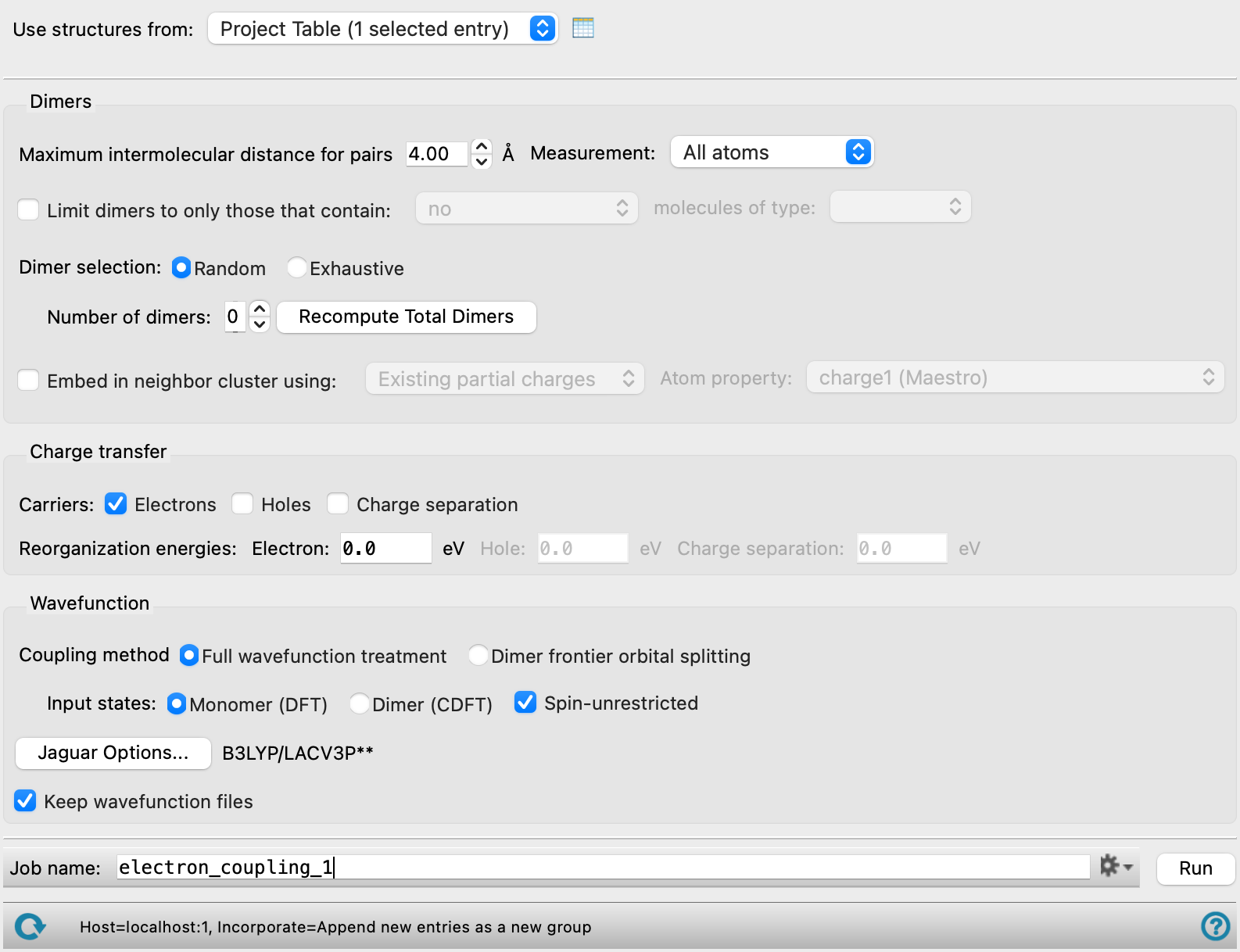
- Dimers section
-
In this section you can select the dimers (donor-acceptor pairs) that are used to calculate the hopping rate.
- Maximum intermolecular distance for pairs box
-
Specify the maximum distance for which two molecules are considered to be a pair. If all designated atoms on one molecule are further than this distance from any designated atom on the other molecule, they are not considered to be a pair. For the center of mass, the distance between the centers of mass determines whether two molecules are a pair, and you will need to choose a value that is related to the size of the molecule rather than the distance between peripheral atoms.
- Measurement option menu
-
Choose whether the measurement of intermolecular distances applies to all atoms (All atoms) to atoms other than hydrogen (Heavy atoms) or to the center of mass (Center of mass). Choosing the heavy atoms option reduces the number of pairs found for a given distance; likewise choosing the center of mass reduces the number of pairs found. You may have to change the Maximum intermolecular distance for pairs value for the center of mass option to a value related to the size of the molecules.
- Limit pairs to only those that contain N molecules of type option menus
-
Restrict the choice of molecules for donor-acceptor pairs. For two molecules to be considered, they must satisfy the conditions specified by the option menu. The "types" are the unique molecular structures in the input. The menu lists the molecular formula and the number of molecules with that formula (and the same connectivity) in the system.
- Dimer selection options
-
These options allow you to select dimers at random or to perform an exhaustive calculation based on all possible dimers that are neighbors (have atoms within the specified maximum distance). Random selection is a compromise between speed and accuracy.
- Number of dimers text box
-
If you choose dimers at random, you can specify the number of dimers to choose in this text box. You must click Recompute Total Dimers to calculate the number of dimers before you can specify the number to choose at random.
- Recompute Total Dimers button
-
Recompute the total number of dimers that meet the criteria specified in this section (distance and limits). The button is replaced by the computed number.
- Embed in neighbor cluster using option and menu
-
Embed the dimer in a set of point charges obtained from the partial charges of the atoms in the neighboring molecules. The neighboring molecules are selected using the same distance criterion as for the dimers. This option allows you to account for the effects of the immediate electrostatic environment of the dimer. Note that SCF convergence can be more challenging when embedding in a set of point charges is used.
There are two choices for the source of the point charges:
-
Existing partial charges—Use the partial charges taken from a Maestro property. The property can be chosen from the Atom property option menu (see below)
-
ESP charges—Use partial charges derived from the electrostatic potential for the neighboring molecules. A single-point B3LYP Jaguar calculation for the ESP charges is run on one molecule of each type in the system as an isolated molecule, and the ESP charges are used for all molecules of that type. You can specify the basis set for the ESP charge calculation in the Basis set text box (see below).
- Basis set text box
-
Jaguar keyword for the basis set used for the ESP charge calculation. When you click in the text box, a small panel is displayed in which you can select the basis set and add diffuse and polarization functions. You can also edit the keyword directly. See Basis Sets for a list of basis set keywords.
- Atom property option menu
-
Choose the atom property that defines the partial charges from the option menu. The option menu is populated with the available charge properties.
-
- Charge transfer section
-
Specify the type of carrier and the reorganization energy.
- Carriers options
-
Select one or more carrier types to use for this material.
- Electrons—electron is transferred from an anion to a neutral molecule.
- Holes—electron is transferred from a neutral molecule to a cation.
- Charge separation—electron is transferred from one neutral molecule to another. If you select this carrier type, the Coupling method and Input states options for dimers are disabled.
- Reorganization energies text boxes
-
Specify the reorganization energy for each type of carrier. You must supply values that are different from zero. You could, for example,use the Optoelectronics Calculations Panel to calculate them. The reorganization energy is assumed to have been calculated for the isolated, ground state relaxed molecule, and is assumed to be independent of the geometry in the mixture. If you have a heterogeneous system, you must decide how to represent the reorganization energy as a single value.
For charge separation, the reorganization energy is not calculated directly from the Optoelectronics Calculations Panel, but it can be calculated from the results as the sum of the Hole Small Polaron Stabilization Energy and Electron Small Polaron Stabilization Energyproperties, which are reported as primary properties in the Project Table.
- Wavefunction section
-
Provide information on generation of the wave function for the electron transfer and reorganization energy calculations. The geometry used for the electron transfer is taken from the input simulation.
- Coupling method options
-
Select an option for the coupling method used to calculate the rate:
- Full wavefunction treatment—Use the wave functions for the species in which the charge is localized on one or the other monomer to calculate the coupling matrix element. You can use either dimer wave functions or monomer wave functions by choosing one of the Input states options.
- Dimer frontier orbital splitting—Use the dimer frontier orbital approximation to calculate the coupling matrix element. In this approximation, the matrix element is calculated from the difference between the LUMO and the LUMO+1 orbital energies for electron transfer, and the difference between the HOMO and the HOMO+1 orbital energies for hole transfer, taken from calculations on the neutral dimer. Not available when the carrier choice includes Charge separation.
- Input states options
-
Choose the type of wave function used for the states that are used to calculate the electron transfer rate. These options are only available for the Full wavefunction treatment coupling method.
- Monomer—Perform calculations on each monomer of a pair, in which the carrier is situated on the monomer. This involves a calculation on each of the monomers with and without the charge, 4 calculations in all. If you select Embed in neighbor cluster using in the Dimers section, the other member of the pair is represented by point charges rather than being absent entirely.
- Dimer—Perform two constrained DFT calculations on the dimer, one in which the carrier is localized on one monomer, and one in which the carrier is localized on the other monomer. Not available when the carrier choice includes Charge separation.
- Spin-unrestricted option
-
Select this option to enable spin-unrestricted (UDFT) treatment of the open shell system created by removing or adding an electron. RODFT calculations are carried out if this option is deselected. Only available for the Full wavefunction treatment coupling method.
- Options button
-
Set Jaguar options for the electron coupling calculations. Opens the Jaguar Options - Electron Coupling Dialog Box. This dialog box also allows you to specify additional Jaguar keywords. The solvent (if any), level of theory, and basis set are shown to the right of the button.
-
If you modify the basis set, you should choose a basis set that has polarization and diffuse functions, if possible, as these are important for calculating the electron transfer rate.
- Keep wavefunction files option
-
Keep the Jaguar restart (.01.in) files, which contain the wave function from the calculation. Keeping them uses more disk space; not keeping them can cause a restart to recalculate results.
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Electron Coupling - Job Settings Dialog Box, where you can make settings for running the job.
- Status bar
-
The status bar displays information about the current job settings and status for the panel. The settings includes the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
Use the Reset button
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel. The status bar also contains the Help button
 , which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a
, which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a  button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.
button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.