Compute KMC Charge Mobility Panel
Compute charge mobility in an amorphous solid or a crystalline solid using kinetic Monte Carlo (KMC) methods.
To open this panel: click the Tasks button and browse to Materials → Quantum Mechanics → KMC Charge Mobility → New Compute KMC Charge Mobility.
To open this panel from the entry group for the results of a job .
.
Please note that as of Schrödinger Suite Release 2025-2 kinetic Monte Carlo calculations are run using new Schrödinger functionality. To access the previous version of the panel using VOTCA [25], disable the KMC_INTERNAL_ENGINE feature flag.
Publications that report results from using VOTCA based kinetic Monte Carlo calculations should cite the VOTCA references [25, 26] as well as references for the Materials Science suite.
The following licenses are required to use this panel: MS Maestro, MS Mobility, Jaguar, QSite
- Using
- Features
- Additional Resources
Using the Compute KMC Charge Mobility Panel
A database containing charge hopping parameter data is required as input for this panel. Such a database can be generated using the Compute KMC Hopping Parameters Panel.
The KMC charge mobility calculations are relatively short and and run serially. In order to reduce the turnaround time, it is recommended that you run on a host with many nodes, so you can distribute the load as much as possible.
The output structure is the same as the input structure but has a number of additional properties. For each applied field, there are three properties that describe the field, named KMC Field Naxis (V/m), and three properties that describe the mobility for each charge carrier, named KMC FNcharge Mobility axis (cm^2/Vs) where N is the index of the field in the list of fields; charge is the carrier type, electron or hole; and axis is the Cartesian axis for the field component, X, Y, or Z. These are the primary properties. These properties are used to plot the charge mobility in the Plot KMC Charge Mobility Panel. In addition, there is a set of secondary properties for the velocity of the charge, named KMC FNcharge Velocity (m/s).
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
There are several possible next steps for a KMC charge mobility job. To choose the next step from a suggested list, including computing (more) mobilities, use the Workflow Action Menu for the results entry or entry group in the Entry List (Entries).
Alternative methods are available for computing charge mobilities, in the Charge Mobility Panel.
Additional references relevant to charge transport in organic semiconductors can be found here [20 - 24].
Compute KMC Charge Mobility Panel Features
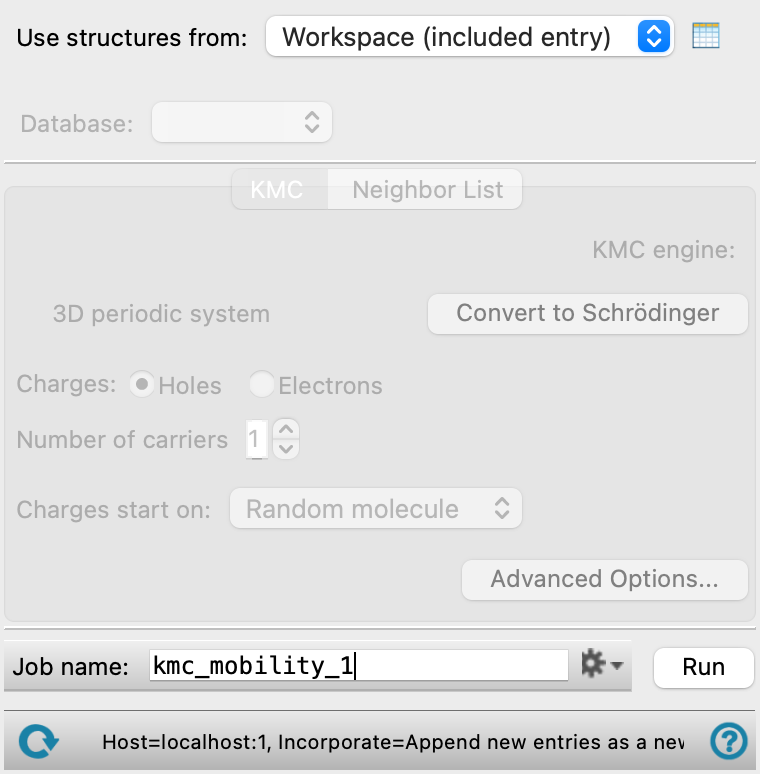
- Use structure from option menu
-
Choose the structure source for computing charge mobilities. If the structure source contains multiple structures, only the first structure is used. The structure must have periodic boundary condition properties.
- Workspace (included entry)—Use the entry that is currently included in the Workspace. Only one entry must be included in the Workspace.
- File—Use the specified file. When this option is selected, the File name text box and Browse button are displayed.
- Database—Use the structure whose identity is recorded in the specified SQL database. When this option is selected, the File name text box and Browse button are displayed, so you can specify the database.
- Open Project Table button
-
Open the Project Table panel, so you can

- File name text box and Browse button
-
Enter the file name in this text box, or click Browse and navigate to the file. The name of the file you selected is displayed in the text box.
- Database menu
-
Select the hopping parameter database from the menu for the charge mobility calculation. This option is present if you choose Workspace (included entry) or File from the Use structures from option menu. Use the Compute KMC Hopping Parameters Panel to generate a hopping parameter database.
- KMC tab
-
Set options for the kinetic Monte Carlo calculations for charge mobility.
- KMC engine text
-
Displays the KMC engine in use, Schrödinger.
- System type text
-
Displays the system type as 3D periodic system or 2D periodic in X and Y.
- Charges options
-
Select the type of charge carrier (Holes or Electrons) for the charge mobility.
- Number of carriers text box
-
Set the number of charge carriers to use in the mobility calculations. If you have more than one charge carrier, the Coulomb interaction option is enabled.
- Coulomb interaction option
-
Use the full bare electrostatic interaction between the charges. The charges are placed at the center of mass of the molecule that has the charge, for the purpose of evaluating this interaction. Only present when the Number of carriers is set to more than one.
- Effective charge text box
-
Specify the effective charge for coulomb interactions. Only present when the Number of carriers is set to more than one and only available when the Coulomb interaction option is selected.
- Charges start on option menu
-
Specify where the injected charges start.
-
Random molecule—the molecules on which the charges start are selected at random from all the molecules in the system.
-
Molecule number—the charge starts on the molecule number specified in the displayed text box. The molecule number is displayed in the status bar when the pointer is over an atom in the Workspace. You can also label molecules with the molecule number (see Labeling Atoms and Bonds). This option is only available if a single charge is injected into the system.
-
Molecule type—the charges start on randomly selected molecules of the type chosen from the option menu that is displayed. This secondary option menu is populated from the molecule types found in the input system.
-
- Advanced Options button
-
Set additional options for the kinetic Monte Carlo calculations. Opens the Compute KMC Charge Mobility — Advanced Options Dialog Box.
- Neighbor List tab
-
Set options for determining which molecules are neighbors.
- Cutoff distance text box and based on option menu
-
Displays the cutoff distance in angstroms, and choose how the distance was calculated in the loaded database. Noneditable.
- Statistics report
-
Reports the average, maximum, and minimum numbers of nearest neighbors, and the total number of neighbor pairs.
- Highlight button
-
Highlight the molecule mentioned in the statistics in the Workspace. The molecule is represented as tubes, and colored red; its neighbors are colored yellow. You might want to color all the molecules with a color that does not obscure this highlighting.
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Compute KMC Charge Mobility - Job Settings Dialog Box, where you can make settings for running the job.
- Status bar
-
The status bar displays information about the current job settings and status for the panel. The settings includes the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
Use the Reset button
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel. The status bar also contains the Help button
 , which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a
, which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a  button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.
button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.