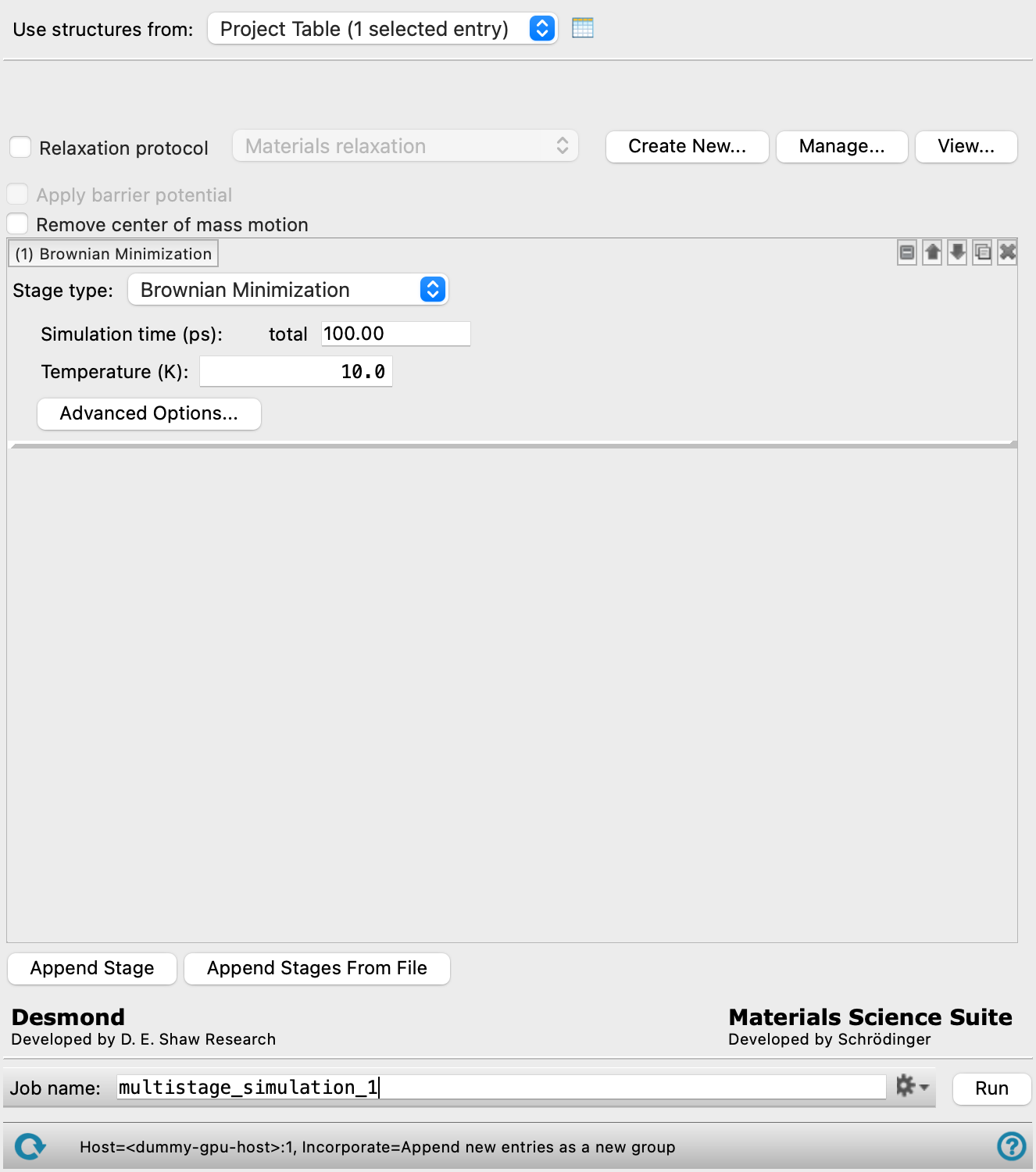
MD Multistage Workflow Panel
Set up a multistage molecular dynamics simulation workflow, consisting of an optional relaxation protocol and an arbitrary number of simulation stages. The results of each stage are fed into the next stage, including the final velocities.
To open this panel, click the Tasks button and browse to Materials → Classical Mechanics → MD Simulations → MD Multistage Workflow.
You can also open this panel with the Workflow Action Menu  for project entries for various jobs that created a Desmond model system. The panel opens with the system in the project entry loaded.
for project entries for various jobs that created a Desmond model system. The panel opens with the system in the project entry loaded.
The following licenses are required to use this panel: MS Maestro, Desmond, OPLS (optional), MS Force Field Applications (optional), MS CG (optional)
- Using
- Features
- Additional Resources
Using the MD Multistage Workflow Panel
The Multistage Simulation Workflow panel uses a Desmond model system as input. The model system can be generated in the Disordered System Builder Panel, or the standard Desmond System Builder panel. It also supports coarse-grained models prepared with the Coarse-Grained Force Field Assignment Panel. Note that checkpoint files (.cpt) are not supported as input.
The panel was primarily designed to allow processing of the initial condensed phase model generated from the Disordered System Builder, using a series of NVE, NVT, and finally target temperature NPT simulations to predict the amorphous system density and structure. However it can be used for any Desmond simulation, and compute properties such as the coefficient of thermal expansion or Tg. With the addition of Analysis stages, you can use the MS MD Trajectory Analysis Panel for analysis of bulk properties from the preceding stages. For example you could run simulations at a range of temperatures and do the analysis at each temperature. The default settings used for the Desmond simulations are different from the Desmond defaults: they have been customized for materials science applications, so you should use this panel in preference to the standard Desmond panels for the same task. In addition, there are some features (such as flexible-angle coupling) that are only available via this panel.
When the panel opens, a single simulation stage is shown below the Model system section. Each simulation stage has controls for a single stage of the simulation. You can add stages in the following ways:
-
Click Append Stage to add a new default stage at the end of the list.
-
Click the Duplicate button
 at the top right of a stage to duplicate the stage, with all its settings. The new stage is placed below the source stage.
at the top right of a stage to duplicate the stage, with all its settings. The new stage is placed below the source stage. -
Click the Append Stages from File button, and import the stages from a previous simulation. A file selector opens so you can locate the desired
.msjfile. You can only import stages from a minimization, molecular dynamics, or simulated annealing run, or a multistage run from this panel. The stages are added at the end of the list.
You can also add a predefined relaxation protocol before the simulation stages, or run it on its own by deleting all the simulation stages.
You can rearrange stages with the arrow buttons 
 at the top right of the stages. This makes it easy to duplicate an earlier stage then move it down into the desired location, and modify the settings.
at the top right of the stages. This makes it easy to duplicate an earlier stage then move it down into the desired location, and modify the settings.
If you want to see only some of the stages, you can show or hide the options with the show and hide buttons 
 . This is useful when you have a number of stages and want to compare two separated stages, for example.
. This is useful when you have a number of stages and want to compare two separated stages, for example.
Note: You must submit the job to a Linux GPU host, as Desmond only runs under Linux on GPUs.
Note: Input systems with greater than 20,000 atoms may run into memory issues during the calculation. Please ensure the GPU has enough memory before running large systems.
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
MD Multistage Workflow Panel Features
- Use structures from option menu
- Open Project Table button
- Structure force field information
- Relaxation protocol option and menu
- Create New button
- Manage button
- View button
- Apply barrier potential option
- Remove center of mass motion option
- Simulation stage
- Append Stage button
- Append Stages from File button
- Job toolbar
- Status bar
- Use structures from option menu
-
Choose the structure source for the multistage workflow. The structures must be prepared Desmond model systems. You can choose multiple structures, and a job that runs the entire workflow is started for each structure.
- Project Table (n selected entries)—Use the entries that are currently selected in the Project Table or Entry List. The number of entries selected is shown on the menu item.
- Workspace (n included entries)—Use the entries that are currently included in the Workspace, treated as separate structures. The number of entries in the Workspace is shown on the menu item.
- Project Table (n selected entries)—Use the entries that are currently selected in the Project Table or Entry List. The number of entries selected is shown on the menu item.
- Open Project Table button
-
Open the Project Table panel, so you can

- Model system section
- Structure force field information
-
Displays information about the loaded structures. If structures of only one force field type are loaded, the text is directly displayed. If structures of more than one force field type are loaded, click the Multiple Forcefields link to view force field information. The information displayed depends on the force field of the structure, the options are:
-
MLFF(model name)—Displays that an MLFF was used in structure preparation and the model used. To learn about MLFF and its model types available see Machine Learning Force Fields.
-
OPLS type(version)—Displays which OPLS force field was used in structure preparation and the software version in which the structure was generated. To learn more about OPLS force fields see OPLS Force Fields.
-
Coarse-Grained(type)—Displays that a coarse-grained structure was prepared and the method used to prepare it.
The structure count for each type of force field is also displayed.
-
- Relaxation protocol option and menu
-
To run a relaxation protocol before starting the simulations, select Relaxation protocol and select the protocol from the option menu. The built-in protocols include protocols for atomistic simulations and for coarse-grained simulations. The menu only shows items for the model system that is loaded; if none is loaded, the protocols for atomistic simulations are shown. The full list of built-in protocols follows:
-
Materials relaxation—Robust protocol for materials science applications. Consists of:
- 20 ps NVT Brownian minimization at 10 K,
- 20 ps NPT Brownian minimization at 100 K,
- 100 ps NPT MD stage at 300 K.
-
Compressive—Useful for compressing systems that are built to a low density, such as from the Disordered System Builder. It uses a high-pressure NPT stage (among other stages) to increase the density of the system. Consists of:
- 100 ps NVT Brownian minimization at 10 K,
- 24 ps NVT MD stage at 300 K,
- 240 ps NVT MD stage at 700 K,
- 24 and 240 ps NPT MD stage at 300 K and 1.01325 bar,
- 10000 ps NPT MD stage at 300 K and 1013.25 bar,
- 10000 ps NPT MD stage at 300 K and 1.01325 bar.
-
Ladder polymer relaxation—Relaxation protocol for ladder polymers, developed based on Larsen et. al. Consists of:
- 100 ps NVT Brownian minimization at 10 K,
- 50 ps NVT MD stages at 600 K and 300 K,
- 50 ps NPT MD stage at 300 K and 0.20265 bar,
- 50 ps NVT MD stage at 600 K,
- 100 ps NVT MD stage at 300 K,
- 50 ps NPT MD stage at 300 K and 0.60795 bar,
- 50 ps NVT MD stage at 600 K,
- 100 ps NVT MD stage at 300 K,
- 50 ps NPT MD stage at 300 K and 1.01325 bar,
- 50 ps NVT MD stage at 600 K,
- 100 ps NVT MD stage at 300 K,
- 800 ps NPT MD stage at 300 K and 1.01325 bar.
-
OPLS5 drude convergence—Relaxation protocol using OPLS5 with polarizability on select chemical groups. It is suggested to confirm if this force field is suitable for the system and property of interest. It is recommended to use OPLS4 for initial relaxation for any newly built structures. Vacuum or void space may cause OPLS5 convergence failure. Consists of:
- 100 ps NVT Brownian minimization at 10 K,
- 12 ps NVT MD stage at 10 K,
- 12 ps NPT MD stage at 10 K,
- 12 ps NPT MD stage,
- 24 ps NPT MD stage.
-
Bulk macromolecule relaxation —Custom protocol for bulk macromolecule relaxation. Consists of
- Brownian minimization stage,
- 24 ps NVT MD stage at 300 K,
- 240 ps NVT MD stage at 700 K,
- 24 ps and 240 ps NPT MD stages at 300 K and 1.01345 bar,
- 10000 ps NPT simulated annealing stage at 1.01325 bar,
- 10000 ps NPT MD stages at 300 K and 1013.25 bar,
- 10000 ps NPT MD stages at 300 K and 1.0125 bar.
-
Relaxation1—A fairly standard Desmond protocol, consisting of:
- 100 ps NVT Brownian minimization at 10 K,
- NVT MD stage at 300 K,
- 200 ps NVT MD stage at 700 K,
- NPT MD stage at 300 K and 1.01325 bar,
- 200 ps NPT MD stage at 300 K and 1.01325 bar,
- 1000 ps NPT MD stage at 300 K and 1.01325 bar,
-
Rigorous compression—Useful for rigorously compressing systems that contain stiff polymers. It uses alternating high-pressure and high temperature NPT stages (among other stages) to increase the density of the system. Consists of:
- 20 ps NVT Brownian minimization at 10 K,
- 20 ps NPT Brownian minimization at 100 K,
- 100 ps NPT MD stage at 300 K,
- 3000 ps NPT MD stage at 300 K and 1000 bar,
- 3000 ps NPT MD stage at 600 K and 1.01325 bar,
- 3000 ps NPT MD stage at 300 K and 1000 bar,
- 3000 ps NPT MD stage at 600 K and 1.01325 bar,
- 3000 ps NPT MD stage at 300 K and 1000 bar,
- 3000 ps NPT MD stage at 600 K and 1.01325 bar,
- 3000 ps NPT MD stage at 300 K and 1000 bar,
- 3000 ps NPT MD stage at 600 K and 1.01325 bar,
- 3000 ps NPT MD stage at 300 K and 1000 bar,
- 5000 ps NPT MD stage at 300 K and 1.01325 bar
-
Semicrystal relaxation 1—Custom protocol for semicrystalline polymer relaxation. This protocol should be used by default. It can fail on certain types of structures; if it does, use the Semicrystal relaxation 2 protocol. Consists of:
- 100 ps NVT Brownian minimization at 10 K with crystal atoms restrained,
- 200 ps NPAT MD stage at 50 K and 1.01325 bar with crystal atoms restrained,
- 200 ps NVT MD stage at 50 K without restraints,
- 50 ps NPT-anisotropic MD stage at 50 K and 1.01325 bar without restraints,
- 5 ns NPT-anisotropic annealing stage from 50 K to 300 K at 1.01325 bar without restraints.
-
Semicrystal relaxation 2—Custom protocol for semicrystalline polymer relaxation, with more degrees of freedom. This protocol should only be used when the Semicrystal relaxation 1 fails, as it tends to produce a less crystalline system. Consists of:
- 100 ps NVT Brownian minimization at 10 K with crystal atoms restrained,
- 200 ps NVT MD stage at 100 K with crystal atoms restrained,
- 200 ps NVT MD stage at 50 K without restraints,
- 50 ps NPT-anisotropic MD stage at 50 K and 1.01325 bar without restraints,
- 5 ns NPT-anisotropic annealing stage from 50 K to 300 K at 1.01325 bar without restraints.
-
Constant pressure—Built-in Desmond relaxation protocol in the NPT ensemble. Consists of:
- 100 ps NVT Brownian minimization at 10 K with solute heavy atoms restrained,
- 12 ps NVT MD stage at 10 K with solute heavy atoms restrained,
- 12 ps NPT MD stage at 10 K with solute heavy atoms restrained,
- 12 ps NPT MD stage with solute heavy atoms restrained,
- 24 ps NPT MD stage without restraints.
-
Constant volume—Built-in Desmond relaxation protocol in the NVT ensemble. Consists of:
- 100 ps NVT Brownian minimization at 10 K with solute heavy atoms restrained,
- 12 ps NVT MD stage at 10 K with solute heavy atoms restrained,
- NVT MD stage without restraints.
-
Martini—Relaxation protocol for coarse-grained modeling with the Martini force field and particle types. Only available for coarse-grained model systems with Martini or Shifted Lennard-Jones force fields. Consists of:
- Brownian minimization stage,
- 0.1 ns NVT stages at 10K and 300K,
- NPT with 20ps barostat relaxation time at 10K and 1.01325 bar for 0.1ns, 300K and10 bar for 2ns, 300K and 1.01325 bar for 0.5ns, using Langevin dyanmics, velocity resampling every 1 ps, and time steps of 1, 1, and 3 fs except for the last stage, which uses 10, 10, and 30 fs.
-
Constant Volume Martini—Relaxation protocol for coarse-grained modeling with the Martini force field and particle types in the NVT ensemble. Only available for coarse-grained model systems with Martini or Shifted Lennard-Jones force fields. Consists of:
- 0.1 ns Brownian minimization stage,
- 0.1 ns NVT stage at 10K,
- 0.5 ns NVT stage at 300K,
- 1.2 ns NVT stage at 300K.
-
Repulsive harmonic—Protocol for dissipative particle dynamics with a repulsive harmonic potential. Consists of:
- 100ps Brownian minimization stage at 10K
- 0.1 ns NVT stages at 10K and 300K with Langevin dynamics, and velocity resampling every 1 ps for the 10K NVT stage.
This option and menu provide a protocol for coarse-grained systems with the Martini force field (or shifted Lennard-Jones potentials). For other coarse-grained systems you will have to design your own protocol. See Setting Up the Coarse-Grained Simulation for more information on appropriate stages to include for relaxation of these systems.
If you just want to run the relaxation protocol, you can delete all other stages in the workflow to run the relaxation job.
-
- Create New button
-
Create a new relaxation protocol from the simulation stages currently defined. These stages include the currently selected relaxation protocol if the Relaxation protocol option is selected. Opens the Create Custom Relaxation Protocol dialog box, in which you can name the protocol. The new protocol is then added to the Relaxation protocol menu, and is stored in your Schrödinger user resources directory. The model system type (atomistic or a coarse-grained type) is included in the information stored, so it is only shown on theRelaxation protocol menu for the same type of system.
- Manage button
-
Manage your custom relaxation protocols. Opens the Manage Custom Relaxation Protocol dialog box, in which you can choose a protocol from an option menu, and delete it by clicking Delete Protocol. The protocol is then removed from the Relaxation protocol menu.
- View button
-
View the stages of the relaxation protocol selected in the option menu. Opens the Relaxation Protocol Details dialog box, in which the stages of the protocol are listed.
- Apply barrier potential option
-
Select this option to take in account any repulsive barriers that have been defined for the system during the simulation. Barriers can be defined from the Set Barrier Potential for MD Panel.
Only available when the input structure is created with the Set Barrier Potential for MD Panel.
- Remove center of mass motion option
-
Select to remove net center of mass motion from the system velocities for the system for all stages of the calculation. Most real systemsin equilibrium mathematically contain no net center of mass motion, but some may arise due to numerical implementation. For infinitely bonded systems, this option selected by default. For all other systems, the option is not selected. For certain systems (e.g. bilayers) and calculations (e.g. diffusion), it is recommended to select this option.
- Simulation stage
-
Each simulation stage has controls for a single stage of the simulation.
- Stage label
-
The label indicates the stage number and the type of the stage. It is updated if the stage type is changed or the stage is moved. If the stage settings are hidden, the label gives a brief summary of the main stage parameters.
- stage management buttons
-
These buttons perform display and ordering operations on the stage. They allow for easy duplication and rearrangement of stages.
Show or hide the contents of the stage. When hidden, only the stage number, label (if any) and these buttons are displayed. This is useful when you have a number of stages and want to compare two separate stages, for example. Move the stage up or down one place in the list.
Duplicate the stage. This is useful for creating similar stages with variations on the settings. 
Delete the stage. - Stage type option menu
-
Select the type of simulation stage, from Brownian Minimization, Molecular Dynamics, MLFF Molecular Dynamics, DPD Molecular Dynamics, Martini Molecular Dynamics, Simulated Annealing, Average Cell, Deform Cell, or Analysis. When you select a stage type, the label is updated with the type, and the relevant settings are displayed below these options.
- Simulation options
-
Set options for the simulation stage. The options depend on the simulation type. The options Molecular Dynamics and Simulated Annealing are the same as in the corresponding Desmond panels, including a button to open the relevant Advanced Options dialog box for advanced settings. The exceptions are that the relaxation options are not present, and there is an additional option for the time step, described below. If you chose multiple structures as input, any options that are related to the atoms in the structures (like restraints) must be specified with expressions that can be applied to all structures.
The MLFF Molecular Dynamics stage has the same options available as the Molecular Dynamics stage.
The DPD Molecular Dynamics and Martini Molecular Dynamics stages have the same options available as the Molecular Dynamics stage, except that the scaled effective solvent settings are not available. The settings may be different from regular MD, as some of them are set from the input coarse-grained structure. These stages should be used for a coarse-grained structure that has been prepared for simulation with the Coarse-Grained Force Field Assignment Panel.
Links to the descriptions are given here:
- Brownian Minimization options
- Molecular Dynamics options
- Simulated Annealing options
- Average Cell options
- Deform Cell options
- Analysis options
- Time step text box
- Set from Structure button
- Brownian Minimization options
-
The Brownian Minimization stage relaxes a system into a local energy minimum, using a Brownian motion simulation.
- Simulation time text box
-
Enter the maximum simulation time for the Brownian motion simulation, in ps.
- Temperature text box
-
Specify the temperature to be used, in kelvin.
- Advanced Options button
- Opens the Brownian Minimization - Advanced Options dialog box, in which you can control many more of the detailed settings for the simulation than are available in the Brownian Minimization options.
- Average Cell options
-
The Average Cell stage constructs a new cell whose dimensions are averages over a specified portion of the end of the simulation from the previous stage. The percentage of the trajectory to use can be specified in the text box. This stage is intended to create an average cell from the end of a stable NPT simulation where the cell volume is fluctuating by a few percent at most. As the fluctuations get bigger, it is more likely that a relaxation stage would be needed before using the cell for the next stage. This stage is useful when a stable cell is needed for NVT simulations, where the cell size is stabilized in a preceding NPT simulation. The final cell size from an NPT simulation might not adequately represent the stable cell, but the average should. The fractional coordinates from the last frame are used to define the structure for the average cell. Please note that an Average Cell stage can follow an Analysis stage, but not precede it.
- Deform Cell options
-
The Deform Cell stage deforms the cell by changing its cell lengths, and optionally changing the atomic coordinates by the same factor. The Deformation vector text boxes specify the factors by which each of the cell lengths (a, b, and c) are multiplied.
The deformation factors are also applied to the coordinates of the atoms if Update atom coordinates is checked. To deform the cell without changing the atom coordinates (thus creating space between adjacent cells in the deformation directions), clear this option.
- Analysis options
-
The Analysis stage performs the analysis and generates the
.eaffile needed to use the MS MD Trajectory Analysis Panel for analysis of bulk properties. You can select the properties to calculate with the check boxes. There is a limit of 15000 molecules for the calculation of properties other than the volume, density, and specific heat capacity (for which there is no limit). By default, all properties are selected for analysis. Specific heat capacity can only be calculated for the NVE, NVT, and NPT ensembles. - Time step text box
-
Specify the time step, in fs. Setting the time step is useful at elevated temperatures, where a smaller value than the default is often needed, and for coarse-grained modeling, where a larger value than the default is useful. This text box is available for Molecular Dynamics, MLFF Molecular Dynamics, and Simulated Annealing simulations.
The values in the Advanced Options Dialog Box for these stages are updated to match: the bonded and near time steps are set to this value, and the far time step is set to three times this value. Similarly, changing the bonded time step (but not the other two) in the Advanced Options Dialog Box changes the value in this text box.
- Set from Structure button
-
Set the time step from the structure, according to the stage.
This button is only present in the DPD Molecular Dynamics and Martini Molecular Dynamics stages. Otherwise, the time step is the one set for the stage.
For the DPD Molecular Dynamics stage, the time step is set from the cg time step (fs) property stored with the structure.
For the Martini Molecular Dynamics stage, the time step is set according to the following formula

where
-
3.0 is the target energy conservation in kelvin per degree of freedom per nanosecond,
-
A = 0.007448,
-
C = −1.387,
-
B = 0.3370,
-
max(k/μ) is the largest ratio of the bond force constant k in kcal mol−1Å−2 to the reduced mass μ of any bond in amu. If there are no bonds, k is set to zero.
-
- Append Stage button
-
Add a stage to the end of the workflow. By default, the stage is a minimization stage with the default minimization parameters.
- Append Stages from File button
-
Add stages to the workflow from a previous simulation, by importing them from a multisim MSJ file (
.msj). Opens a file selector, in which you can navigate to and open the file. The simulation must be one that was initiated from this panel, or from the Minimization Panel (Desmond → Minimization), the Molecular Dynamics Panel (Classical Simulation → Molecular Dynamics), or the Simulated Annealing Panel (Classical Simulation → Simulated Annealing). Importing stages from other types of jobs or types of model systems (atomistic or a coarse-grained type) is likely to cause failure, and a warning is posted for any stage that is not recognized by this panel. It is highly recommended that you import simulations that were initiated from this panel as these simulations have settings that were customized for materials science applications.MD stages are converted to DPD MD stages on input if the MD stages were written out with a DPD thermostat.
If you import a minimization stage that was run prior to the 2019-1 release, the stage is converted to a Brownian Minimization stage (which is the same as the former Brownian Dynamics stage).
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the MD Multistage Workflow - Job Settings Dialog Box, where you can make settings for running the job.
- Status bar
-
The status bar displays information about the current job settings and status for the panel. The settings includes the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
Use the Reset button
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel. The status bar also contains the Help button
 , which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a
, which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a  button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.
button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.
Tutorials
- Building, Equilibrating and Analyzing Amorphous Polymers
- Building a Semicrystalline Polymer
- Solid Electrolyte Interphase Calculations
- Building and Analyzing a Complex Lipid Bilayer and Embedding a Membrane Protein
- Disordered System Building and Molecular Dynamics Multistage Workflows
- Nanoemulsions with Automated DPD Parameterization
Videos