Coarse-Grained Modeling with the Martini Force Field
This document outlines the workflow for coarse-grained modeling using the Martini force field.
The following topics are covered in the sections below.:
- Coarse Grained Sketcher
- Disordered System Builder
- Coarse-Grained Force Field Assignment
- Multistage Relaxation and Simulation
Coarse-Grained Sketcher
It is important to distinguish between Martini force field site types (Cx, Qx, Px, Nx) and sites within a molecule, e.g., BUT (-CH2-CH2-CH2-CH2-, which can be linked together to form chains of different lengths and can when needed be consisted to include capping H atoms). The sketcher generally uses site types within a molecule. While you can sketch using entirely new site types when drawing in a molecule, you will eventually need to select the Martini site types and valence parameters. Using known predefined site types in the sketcher allows the Martini types and many or all of the valence parameters to be based on known selections and obtained from an existing force-field database. It is worth noting that site types within a molecule can be used more generally than within a specific molecule. For instance, the sites within the phosphatidyl choline head group from DPPC (namely, DPPC_NC3 - DPPC_PO4 - DPPC_GL - DPPC_GL) can used in other phosphatidyl choline lipid molecules.
There are many sites within a molecule that are predefined for Martini and accessible in the Coarse-Grained Sketcher. For a list of known types please see Site Types for Martini. In addition, if you want to use one of the molecules that is actually described on that page (e.g., DPPC), many of them can be directly imported into Maestro from the distribution using the Import Martini Coarse-Grained Structures Panel.
The Coarse-Grained Sketcher Panel is easy to use. The help page for this panel describes the steps needed for sketching particles. For Martini force fields, if you want to use some of the known site types within a molecule, start by setting Use particle types from to Force field file and click Create. In the Create Particle Types from a Force Field File Dialog Box, if you have not already made a custom Martini force field file, set the Location to Installation and set Select force field file from which to create particle types to Martini (the default). Then proceed to select and add the particle types as described in the help page.
The sketching itself is fairly intuitive. Click on the desired site type (hover over it with the mouse to get a reminder about what type it is) and then start drawing the molecule in the Sketcher's workspace. When you are done drawing in the molecule, give it a name in the Title text box and click Create Project Entry.
Below is an example of a moderately challenging molecule (PAMAM) from the literature (Lee and Larson, J. Phys. Chem. B, 2008, 122, 7778).
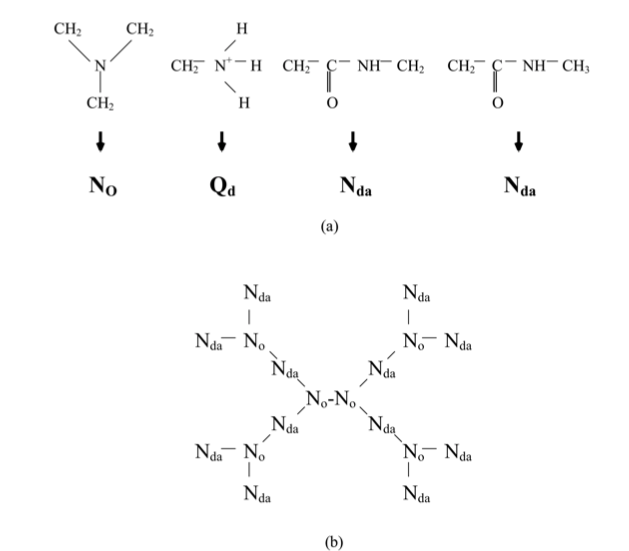
Below are the corresponding PAMAM site type names within a molecule.
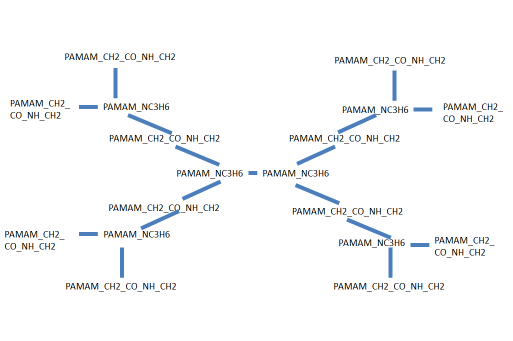
The corresponding model of PAMAM in the Coarse-Grained Sketcher looks like this:
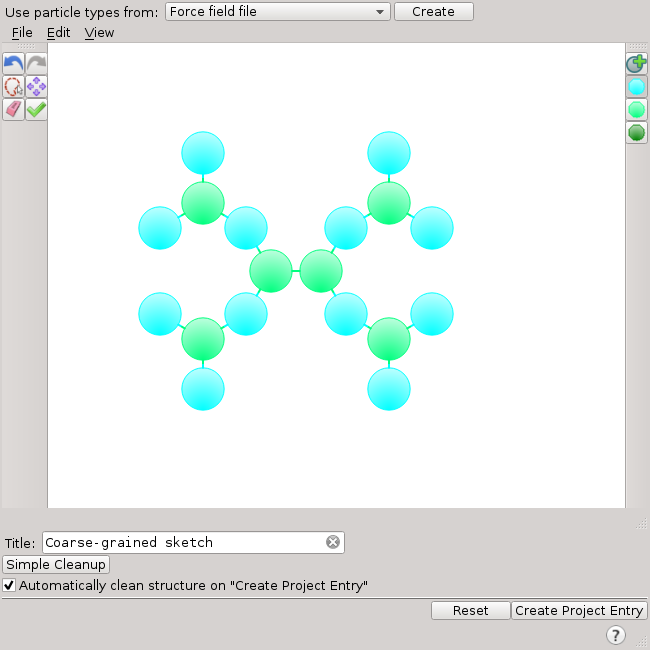
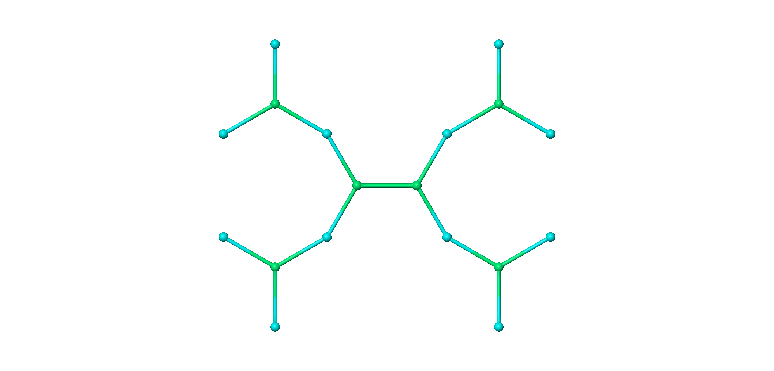
After clicking Create Project Entry, the PAMAM molecule in the Maestro Workspace looks like this:
Disordered System Builder
Once there are entries for each type of coarse-grained molecule needed for the study in the Project Table, the Disordered System Builder can be used to construct a model system of the desired composition. First select the entries needed and then open the Disordered System Builder Panel. Start by setting the total number of molecules first (usually more than about 1,000 coarse-grained sites are needed) and then set the number of molecules for each component (type of molecule). The following non-default settings are recommended:
-
For Periodic boundary conditions select Create new cubic PBC.
-
Under Disorder select Tangled chain for Initial state if the molecules are simple and linear, otherwise select Amorphous.
-
In the Disorder Options Dialog Box: (click Disorder Options to open):
-
Set the Initial density to 0.3
-
Increase Placement attempts per molecule to 128.
-
Turn off Color molecules by component for multicomponent systems, to retain the coloring by functional group for polyfunctional molecules (unless you want molecules colored uniformly by type).
-
Click OK.
-
Click Run to start the job. Many Disordered System Builder jobs for molecules that are compact and consist of a few hundred small molecules take only a few minutes to complete. Larger systems, particularly with long flexible molecules can take significantly longer.
Coarse-Grained Force Field Assignment
Assignment of the force-field parameters is done in the Coarse-Grained Force Field Assignment Panel. This section discusses the ways of specifying site types in this panel:
- Use of Standard Site Types Within a Molecule
- Specifying Standard Martini Site Types
- Specifying Custom Martini Site Types
Regardless of which strategy is used once the parameters are entered, you can save a local copy of the parameters for future use by entering a Force field name (and an optional description) and clicking the Save button, before clicking the Run button. Clicking the Run button assigns the force field parameters and creates a new entry in the Project Table.
Use of Standard Site Types Within a Molecule
If you used known site types within a molecule when creating a molecule in the Coarse-Grained Sketcher, then assigning force-field parameters is easier or even trivial. First, you should read in the corresponding force-field file using the Import force field controls at the top of the panel:
-
Select the location: Installation or Local.
-
The installation currently only contains a standard Martini file. However this is a great place to start if you have not previously saved relevant parameters.
-
Local only contains force field files if you have previously saved them.
-
-
Select the force field to import (usually just Martini)
-
Click the Import button
If you have used known site types within a molecule:
-
The corresponding Martini site types for that molecule are filled in automatically in the Site tab
-
For known types, bonds, angles, improper torsions, and torsions involving these sites within a molecule, the parameters are filled in automatically. Note, for combinations of sites not covered by the force field file, you still need to add valence terms with the appropriate parameters.
-
Many non-bonds involving these types interacting among themselves or with other sites of a standard Martini type are filled in automatically in the Nonbond tab. Nonbonded interactions involving "custom" Martini types are filled in using default parameters unless they are listed in the imported force field file. See the custom site type section below for more information.
If all molecules in the system are represented in the force-field file and if those site types within a molecule are used to draw the molecules in the coarse grain sketcher, then simply selecting and importing that force-field file (at the top of this panel) results in all of the parameters being filled in (you may need to go to the angle and torsion tabs and click Enumerate Types to fill them in). You then need only to click Run to complete the process.
Specifying Standard Martini Site Types
You can still import a Martini force-field file even if it does not cover all of the sites within a molecule needed for the current system. Consulting the document Selecting Martini Parameters should help with this process.
If at least some site types within a molecule were not specified then:
-
You should select an appropriate site type in the Site tab (perhaps after consulting the main Martini paper1).
-
The nonbonded interactions amongst all site types within a molecule involving standard types or between types covered by a force-field file, if one was read in, are filled in automatically. Nonbonded interactions involving "custom" Martini types are filled in using default parameters unless they are listed in the imported force-field file. See the custom site type section below for more information.
-
Bonds involving the sites for which Martini Site types were selected will be listed automatically, however, their parameters will be initially set to default values which may not be appropriate.
-
Angles, dihedrals, and improper dihedrals involving the sites for which Martini Site types were manually selected will not automatically appear in the corresponding tabs and when they are added will be assigned default parameters which in most cases should be adjusted.
Specifying Custom Martini Site Types
You can still import a Martini force field file even if it does not cover all of the sites within a molecule needed for the current system, and custom sites will be used.
Many extensions to the basic Martini force field[1] involve Martini site types that are not part of the standard set (e.g., PEO_SP0 for -CH2-O-CH2-). In some cases these site types within a molecule are available in a force-field file. If so, their use is similar to a standard Martini type except that typically not all of the parameters for their nonbonded interactions with other known Martini types will be predefined and default parameters will initially appear in the table. You should check the Nonbond tab for interactions involving these sites:
-
These parameters appear with a "G" for guess.
-
Default sigma (4.7 Å) and epsilon (1.0 kcal/mol) values are applied.
-
Under most circumstances these should be adjusted to more suitable values.
-
Valence interactions (bonds, angles, dihedrals and improper dihedrals) involving custom sites are handled in the same manner as if you selected a standard Martini site in the Site tab and in most cases will need attention, unless they explicitly appear in the force field file.
Multistage Relaxation and Simulation
Coarse grained simulations for materials should be set up using the MD Multistage Workflow Panel. With this panel open and with the an entry in the Workspace that was created by the Coarse-Grained Force Field Assignment Panel, click the Load button. The panel should automatically recognize that this is a system that has Martini force field parameters. Normally, coarse-grained systems are assembled with the molecules packed at a density that is lower than desired. As well, initial packings of molecules (e.g., by the Disordered System Builder) have van der Waals clashes. As a result, relaxation of the system prior to simulation is desirable and usually necessary. There is a pre-defined multi-state relaxation protocol for Martini systems which can be enabled by checking the Relaxation protocol check box.
You can also define a post-relaxation stage to simulate at the desired conditions for a while, before performing a longer production simulation. For this you would generally use the same conditions as the production simulation by selecting the following:
-
Set the Stage type to Martini Molecular Dynamics.
-
Set the simulation time (default 1.2 ns) and temperature (default 300K).
-
Set the time step. Typically, a time step of 10-30 fs is OK with Martini models, depending on the stiffest bonds. Normal strength Martini bonds (12.5 kJ mol−1 Å−2 or 2.988 kcal mol−1 Å−2) support 30 fs.
-
Click Advanced Options.
-
In the Ensemble tab:
-
Select either Langevin or Martyna-Tobias-Klein dynamics as the thermostat and barostat methods.
-
Set a thermostat relaxation time of 10 ps (default) and a barostat relaxation time of 50 ps (default).
-
-
In the Misc tab:
- Be sure that the resampling of the velocities is off (interval 0.0).
You can select the machine or queue to run the job on in the Job Settings Dialog Box, which you open by clicking the gear button beside the Job name text box. Click Run to start the relaxation/simulation workflow.
The production simulation itself can be tacked onto the end of the relaxation protocol or run standalone. The latter permits you to inspect the relaxed system prior to running a longer simulation. If you are doing a standalone production run, load the structure produced by the relaxation protocol into the MD Multistage Workflow Panel, then set up and run the job with the steps given above for setting up a post-relaxation stage.
[1] The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations, Siewert J. Marrink et al, J. Phys. Chem. B 2007, 111, 7812-7824.