Coarse-Grained Force Field Assignment Panel
Assign force-field parameters to a coarse-grained model and create the Desmond CMS file for running a molecular dynamics simulation with this model. The model system is added to the project and displayed in the Workspace.
To open this panel: click the Tasks button and browse to Materials → Classical Mechanics → Coarse Grain Models → Coarse-Grained Force Field Assignment.
The following licenses are required to use this panel: MS Maestro, MS CG
- Using
- Features
- Additional Resources
Using the Coarse-Grained Force Field Assignment Panel
To assign site, bond, or nonbonded parameters:
- Display the tab for the parameter type.
- Choose the instance of the parameter type from the option menu in the Type column for a parameter.
- Edit the cells for the parameters you want to change.
To assign angle or dihedral parameters:
- Display the tab for the parameter type.
- Use the menu in the Type column to choose the type of parameter.
- Edit the cells for the parameters you want to change.
- Click Add Row to add a new parameter type, and repeat steps 2 and 3.
- Repeat for as many parameters as you want to set.
You can save the values for any parameter type in a database, which is kept in your Schrödinger user resources directory. The database is read and the values used as the defaults for any parameters in the current model that are in the database.
We recommend that particles in the system have no more than double the radius of another particle. If particles are vastly different in size, a warning message appears when you run the job, and you must click Continue in order to proceed.
Coarse-Grained Force Field Assignment Panel Features
Each tab consists of a table in which the parameters can be defined. The parameters have an icon with a colored background next to them that indicates the source of the parameter:
- G: guess, orange
- A: average over all occurrences, yellow
- D: taken from the database, blue
- E: edited, green
- H: hidden/encrypted, purple
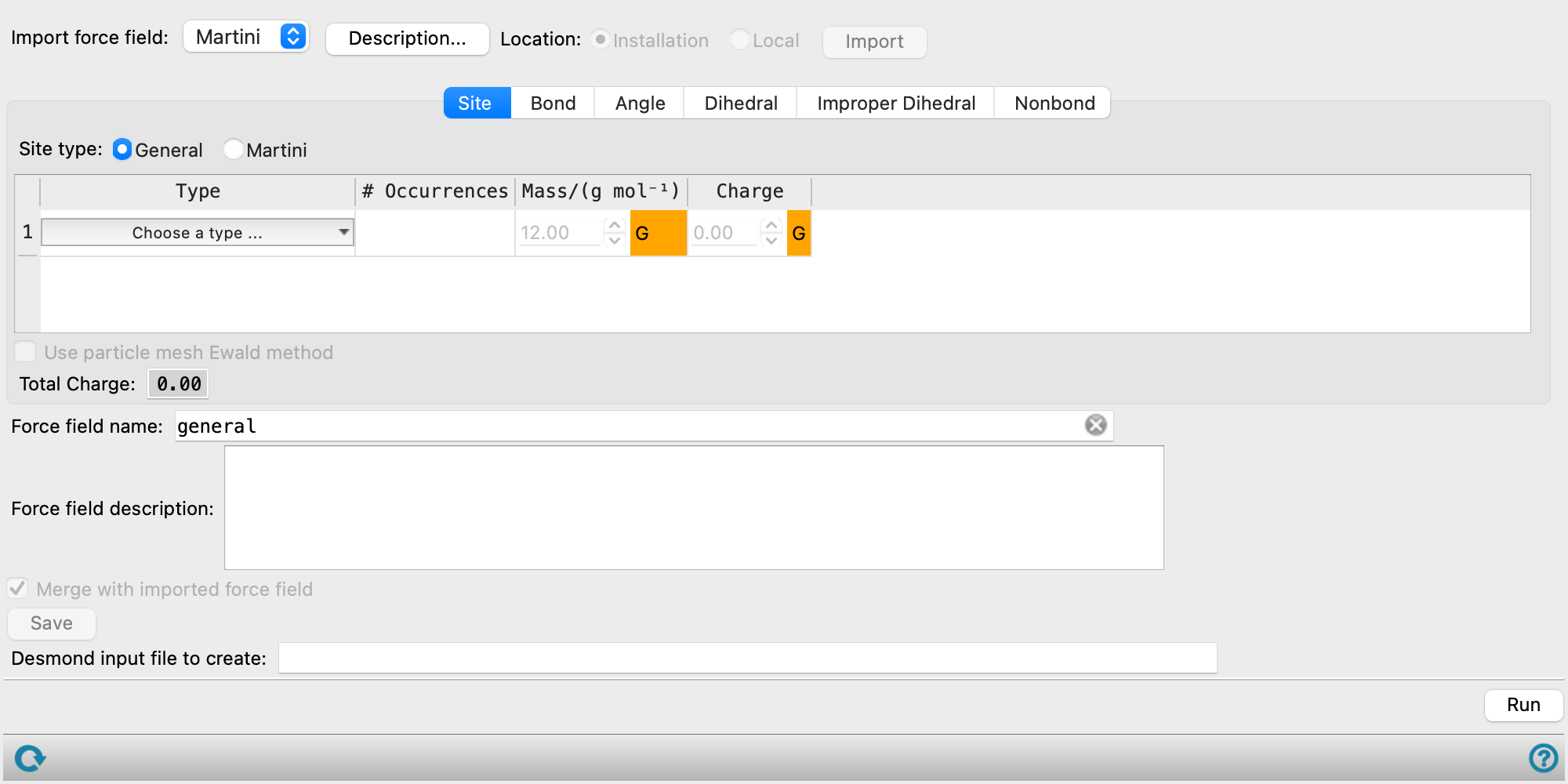
The parameters are assigned from the database, if available, otherwise a guess is made. If encrypted parameters are used in the force field, the panel shows random or invalid values for those parameters.
- Import section
- Site tab
- Bond tab
- Angle tab
- Dihedral tab
- Improper Dihedral tab
- Nonbond tab
- Force field name text box
- Force field description text area
- Merge with imported force field option
- Save button
- Desmond input file to create text box
- Status bar
- Run button
- Import section
-
Import a previously defined force field, from your user resources area or from the installation. The option menu contains the names of the available force fields. Click the Description button to show the description of the force field. Click Import to import the force field. The name and description are shown in the text boxes below the tabs.
- Import force field option menu
-
Choose the coarse-grained force field for import. The choices depend on the Location option selected. The installation contains the Martini and Martini_solution force fields. The menu is populated with the coarse-grained force fields you have saved if you choose Local for the Location.
- Description button
-
Display a description of the chosen force field in a separate panel.
- Location options
-
Select an option for the location of the coarse-grained force field. The force fields listed on the Force field option menu depend on this choice.
- Installation—use the force fields in the installation. These are the Martini, Martini_solvation, or Martini_full force fields [15].
- Local—import a force field from your local user resources directory. These are force fields that you have saved for your own use.
- Import button
-
Import the chosen force field. The panel is set up with options for this force field, and parameters are assigned for the particle types defined in the force field.
- Site tab
-
Specify parameters that apply to individual particles. The controls are displayed in a table, which has rows for each site.
- Site type options
-
Choose the type of sites in the force field.
- General—sites are general-purpose, and the force field is determined by the parameters you set (either by default or explicitly).
- Martini—sites are of the types defined in the Martini force field [15]. If you choose this option, a column for selecting the Martini type is displayed in the table. The Martini parameters are set for the Martini site types when you import the Martini force field. If you have added your own site (particle) types, you can select Custom as a Martini type. Default values will be assigned for bonds and van der Waals parameters between these and other sites, but not for angles. It is good practice to examine and, in most cases, modify all such parameters.
- Site table
-
Define each site by making settings in this table.
Type Type of particle, as defined in the coarse-grained model structure. # Occurrences Number of occurrences of this particle in the model structure. Mass/(g/mol) Mass of the particle. If the model came from the Map Atoms to Particles Panel, the mass is calculated from the constituent atoms, so you should not need to change it. Likewise, if you used the Coarse-Grained Sketcher Panel, the mass set in the panel is used. Charge Charge of the particle. If the model came from the Map Atoms to Particles Panel, the charge is obtained from the formal charges on the atoms in the particle. Martini Type If the sites are Martini sites, choose the Martini type for the particle. The nonbonded parameters for this particle are set according to the Martini force field. See https://cgmartini.nl/docs/downloads/force-field-parameters/martini2/particle-definitions.html or Reference 15 for more information. - Use particle mesh Ewald method option
-
Electrostatic interactions are calculated with the particle mesh Ewald (PME) method when using a Martini force field instead of cutoff parameters. Only available if Martini is selected as the Site type, since the PME method is applied by default for the other coarse-grained force fields.
- Bond tab
-
Specify force-field parameters for the bonds between particles. The controls are displayed in a table, which has rows for each bond. To add a new row to the table, click Define New Bond Type.
Type Bond type, represented as particle1-particle2. All bond types in the model are listed automatically. # Occurrences Number of occurrences of this bond in the model structure. Req/Å Equilibrium bond distance. This is calculated as an average over each occurrence, by default. k/(kcal/mol)/Å2 Harmonic force constant for the bond. The potential is V = k(r−req)2 for general sites, V = (k/2)(r−req)2 for Martini sites. - Angle tab
-
Specify force-field parameters for the angles between particles. The controls are displayed in a table; rows can be added by clicking the Add Row button.
- Enumerate Types button
-
Analyze the structure to enumerate the different types of angles, to populate the option menu in the Type column. As this operation can be slow, the option menu is not populated by default.
- Potential options
-
Choose the type of bending potential. You can only make settings for one type of potential at a time; to use different potentials, you can save the force field and reimport it to add potentials of a different type.
- Harmonic—use a harmonic potential in the bond angle. The potential is V = k(θ − θeq)2 for general sites, V = (k/2)(θ − θeq)2 for Martini sites.
- Trigonometric—use a potential that is quadratic in the cosine of the bond angle. The potential is V = k(cos θ − cos θeq)2 for general sites, V = (k/2)(cos θ − cos θeq)2 for Martini sites.
Note that only one type of bending potential can be set in this tab at a time. However, you can save a force field with one type of potential, make settings with the other type, and then merge the new settings into the saved force field with the Merge with imported force field option.
- Potential definition table
-
Set the angle and force constant of the potential terms in this table:
Type Type of angle. Click the Choose a type button to select the angle type from a menu of particle types. The type is represented as particle1-particle2-particle3, on the menu, when you have made a choice. This menu is populated when you click Enumerate Types; it is not populated by default. # Occurrences Number of occurrences of this angle in the model structure. θeq Equilibrium bond angle, in degrees. This is calculated as an average over each occurrence, by default. k Harmonic force constant for the chosen potential. The units are kcal mol−1 rad−2 for the harmonic potential and kcal mol−1 for the trigonometric potential. Delete Click the button to delete this row from the table.
- Dihedral tab
-
Specify force-field parameters for the dihedral angles between a set of particles. The controls are displayed in a table; rows can be added by clicking the Add Row button.
- Enumerate Types button
-
Analyze the structure to enumerate the different types of dihedral angles, to populate the option menu in the Type column. As this operation can be slow, the option menu is not populated by default.
- Potential options
-
Choose the type of potential. The same form is used for both Martini and general sites. You can only make settings for one type of potential at a time; to use different potentials, you can save the force field and reimport it to add potentials of a different type.
- OPLS—The potential is a sum of terms of the form (Vn/2)(1 − (−1)n cos nφ), with n = 1-4. Vn is the depth of the well (peak-to-trough).
- Trigonometric—The potential is a sum of terms of the form kn(1 + cos(nφ − φeqn)), with n = 1-4. Note that the minimum is in a different place for each n value, in general, so θeqn must be set accordingly.
- Potential terms table
-
Set the angles and force constants of the potential terms in this table:
Type Type of dihedral. Click the Choose a type button to select the dihedral angle type from a cascading menu of particle types. The type is represented as particle1-particle2-particle3-particle4, on the menu, when you have made a choice. This menu is populated when you click Enumerate Types; it is not populated by default. # Occurrences Number of occurrences of this dihedral in the model structure. Vn/(kcal/mol) Well depth for the nth term of an OPLS dihedral potential, in kcal/mol kn/(kcal/mol) Force constant for the nth term of a trigonometric dihedral potential, in kcal mol−1 rad−2. Delete Click the button to delete this row from the table.
- Improper Dihedral tab
-
Specify parameters for improper dihedral angles between a set of particles. The potential is a simple harmonic potential in the angle, V = k(φ−φeq)2. The same form is used for both Martini and general sites. The controls are displayed in a table; rows can be added by clicking the Add Row button.
- Enumerate Types button
-
Analyze the structure to enumerate the different types of improper dihedral angles, to populate the option menu in the Type column. As this operation can be slow, the option menu is not populated by default.
- Potential definition table
-
Set the improper dihedral and force constant of the potential terms in this table:
Type Type of improper dihedral. Click the Choose a type button to select the improper dihedral type from a menu of particle types. The type is represented as particle1-particle2-particle3-particle4, on the menu, when you have made a choice. This menu is populated when you click Enumerate Types; it is not populated by default. # Occurrences Number of occurrences of this improper dihedral in the model structure. θeq/° Equilibrium value of the improper dihedral angle. k/(kcal/mol)/rad2 Harmonic force constant for the angle. Delete Click the button to delete this row from the table.
- Nonbond tab
-
Specify nonbonded interactions between the particles.
- Potential options
-
Choose the type of nonbonded interaction. Settings for each type of interaction are displayed when you make a choice.
- Lennard-Jones—Use a Lennard-Jones type potential for the nonbonded interaction, of the form Cε[(σ/r)m − (σ/r)n], where C is chosen such that ε is the well depth; σ is the value of r where the potential crosses the axis. You can use the same thermostats and barostats as for all-atom simulations.
- Repulsive harmonic—Use a repulsive harmonic potential of the form a (r−c)2 for r < c, 0 for r> c, where c is the cutoff distance. You should use a constant volume ensemble with either a Langevin or a DPD (dissipative particle dynamics) thermostat for the temperature for this type of potential. Note that the DPD thermostat can only be used on a GPU.
- Shifted Lennard-Jones—Use shifted Lennard-Jones potentials for the nonbonded interaction, as used in the Martini force field (see http://www.cgmartini.nl/index.php/faq). Only the ε and σ values may be changed; the latter to one of 4.1, 4.3, 4.5, 4.7, 5.7, and 6.2 as required in the Martini force field. The remaining parameters should be left as they are. An error message is posted if you do make changes to other parameters. The values of ε and σ are pre-populated according to the Martini types set in the Site tab, so you should not need to change them.
You cannot mix Lennard-Jones (of either type) with repulsive harmonic nonbonded interactions in the same simulation: you must choose one or the other for all particles in the simulation.
- Lennard-Jones settings
-
Set the cutoff distance for the Lennard-Jones and Shifted Lennard-Jones potentials.
- Cutoff distance text box
-
Cutoff distance, beyond which the potential is set to zero. This distance should be considered part of the force-field assignment, as it influences the parameters saved in the CMS file. These cutoffs should be used in all subsequent related simulations. It is inadvisable to change the cutoff distance to values other than 12 for the Martini force field.
- Repulsive harmonic settings
-
Set parameters for the repulsive potential.
- Reduced density text box
-
Specify the reduced density (desired number of particles in a box the size of the cutoff distance).
- Cutoff distance text box
-
Specify the cutoff distance for the potential (the value of c in the expression above).
- Density and Lattice scale factor text boxes
-
Displays the real density for the system and the scaling factor for scaling the distances in the structure. These values are calculated from the Reduced density and Cutoff distance. Noneditable.
- Dielectric constant text box
-
Dielectric constant for the system. If water is modeled as a sphere, for example, it has no dipole moment, so a dielectric constant is needed to model the interaction with the water.
- Exclude option and menu
-
Specify which geometric parameters should not be handled by this nonbonded potential. The options are:
- Bonds
- Bonds and 1-3 angles
- Bonds, 1-3 angles and 1-4 dihedrals
- Also exclude pairs within rings and fused-ring systems option
-
Exclude nonbonded pairs that are in the same ring system from this nonbonded potential.
- Parameter table
-
Specify the parameters in this table, which has rows for each particle pair. The table columns depends on the potential chosen. For Lennard-Jones potentials (including shifted potentials), the columns are:
Type Nonbonded interaction type, represented as particle1-particle2. All types in the model are listed automatically. # Occurrences Number of occurrences of this particle pair in the model structure. m exponent, n exponent Exponents of r in the potential, where r is the interparticle distance. Not adjustable for shifted Lennard-Jones potentials. ε/(kcal/mol) Depth of the potential well. σ/Å Scaling parameter for the distance, distance where the potential crosses the axis. For repulsive harmonic potentials, the columns are:
Type Nonbonded interaction type, represented as particle1-particle2. All types in the model are listed automatically. # Occurrences Number of occurrences of this particle pair in the model structure. a/(kcal/mol)/Å2 Harmonic force constant for the bond.
- Force field name text box
-
Enter a name for the force field. When a force field is imported, its name is displayed here. The name is used on the Import force field option menu and on coarse-grained force-field option menus in other panels.
- Force field description text area
-
Enter a description of the force field. When a force field is imported, its description is displayed here, and can be edited. In any panel that allows selection of a coarse-grained force field, the description can be shown by clicking the related Description button. The description given here should therefore be detailed enough to identify the force field for later use.
- Merge with imported force field option
-
Merge any changes to an imported force field into the force field when saving. If not set, the original force field is overwritten with the settings in the panel, and any values in the original force field that are not used by the current system are lost.
This option is useful when you want to include both harmonic and trigonometric potentials for angles or dihedrals, as you can only set one kind of potential at a time. Merging with the imported force field allows you to set different potentials in the panel than the ones in the force field, and merge them to create a mixed potential type force field.
- Save button
-
Save the force field in your user resources area. Unedited hidden force field parameters remain encrypted in the new file, however, hidden parameters which were edited are unencrypted in the new file.
- Desmond input file to create text box
-
Specify the name of the Desmond CMS file to create for the coarse-grained model with the force field.
- Run button
-
Run the job to create the Desmond CMS file. The resulting model system is incorporated into the project and displayed in the Workspace. You can then use it for MD simulations, for example with the Desmond panels or the MD Multistage Workflow Panel.
-
When the panel is run for a force field in which hidden parameters were edited, those parameters become unencrypted in the created force field file and Desmond CMS file.
- Status bar
-
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel.If you can submit a job from the panel, the status bar displays information about the current job settings and status for the panel. The settings include the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
The status bar also contains the Help button
 , which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.
, which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.
Tutorials
- Ibuprofen Cyclodextrin Inclusion Complexes with the Martini Coarse-Grained Force Field
- Building a Coarse-Grained Surfactant Model with Martini Force Field
- Building a Coarse-Grained Polymer Model using Dissipative Particle Dynamics
- Building a Coarse-Grained Skin Model using Martini Force Field
- Nanoemulsions with Automated DPD Parameterization