Semicrystalline Polymer Builder Panel
Build a semicrystalline polymer, consisting of crystalline and amorphous layers.
To open this panel: click the Tasks button and browse to Materials → Structure Builders → Semicrystalline Polymer.
The following licenses are required to use this panel: MS Maestro, OPLS (optional), MS Force Field Applications (optional)
- Overview
- Using
- Features
- Additional Resources
Overview of Semicrystalline Polymer Builder
The process of chain building is the same as in the Polymer Builder Panel for amorphous, tangled chains. However, here we also build the crystalline phase along with the amorphous phase. The process of building a semicrystalline polymer starts with a polymer crystal structure, which defines the polymer type and monomer unit, and is used to define the crystalline layer and grid positions of atoms in the crystal layer. An amorphous region is defined above the slab of the crystalline polymer. These two regions together define the simulation cell for the semicrystalline polymer. By default the simulation cell is orthorhombic, but if you start with a triclinic cell, you can choose to make the output cell triclinic.
The polymer chain building starts at the amorphous layer (lamella), like the tangled chains builder. If the chain touches the crystal surface, i.e. comes within the specified lamella connection distance of the surface, and the remaining chain length is longer than the crystal thickness, then the chain attempts to enter the crystal. When the chain enters the crystal, the subsequent atoms in the chain are placed in the crystal grid positions associated with the chain where it enters, until the other end of the crystal surface is reached. After reaching the other crystal surface, the chain building continues in the amorphous layer until the chain touches the crystal surface again. This process of chain building continues until the specified length and number of polymer chains is reached. Not all crystal chains are necessarily used in this process. The unused chains are deleted, resulting in some voids in the crystal. The percentage of chains used is called the "crystal coverage".
The crystallinity of the final structure (percentage that should be crystalline) is determined by computing the local order parameter (LOP) of all carbon atoms. For each carbon atom, the bond order parameter between that atom (i) and all other carbon atoms (j) within a radius of 6 angstroms is calculated by:

where  and
and  are the unit orientation vectors of carbon atoms i and j connecting the midpoints of the atom's two adjacent backbone bonds. The bond order parameters are averaged to give the LOP for the carbon atom. Any carbon atom with a LOP of 0.73 or more is considered crystalline.
are the unit orientation vectors of carbon atoms i and j connecting the midpoints of the atom's two adjacent backbone bonds. The bond order parameters are averaged to give the LOP for the carbon atom. Any carbon atom with a LOP of 0.73 or more is considered crystalline.
Please cite the appropriate references from the list below in all work that makes use of the supplied crystal structures.
| Polyethylene (PE) | Miao, M. S.; Zhang, M.-L.; Doren, V. E. V.; Alsenoy, C. V.; Martins, J. L. J. Chem. Phys.2001, 115 (24), 11317–11324. |
| Nylon-6 | Holmes, D. R.; Bunn, C. W.; Smith, D. J. The Crystal Structure of Polycaproamide: Nylon 6. J. Polym. Sci.1955, 17 (84), 159–177. |
| Polyvinyl alcohol (PVA) | Bunn, C. Crystal Structure of Polyvinyl Alcohol. Nature 1948, 161 929–930. |
| Polytetrafluoroethylene (PTFE) * | Nakafuku, C.; Takemura, T. Crystal Structure of High Pressure Phase of Polytetrafluoroethylene. Jpn. J. Appl. Phys.1975, 14 (5), 599–602. |
| Poly(ether-ether-ketone) (PEEK) | Fratini, A.; Cross, E.; Whitaker, R.; Adams, W. Refinement of the Structure of PEEK Fibre in an Orthorhombic Unit Cell. Polymer1986, 27 (6), 861–865. |
| Polyvinyl fluoride (PVF) * | Golike, R. C. Symmetry and Dimensions of the Crystalline Unit Cell of Polyvinyl Fluoride. J. Polym. Sci.1960, 42 (140), 583–584. |
| Poly(vinylidene fluoride) (PVDF) | Li, M.; Wondergem, H. J.; Spijkman, M.-J.; Asadi, K.; Katsouras, I.; Blom, P. W. M.; Leeuw, D. M. D. Revisiting the δ-Phase of Poly(Vinylidene Fluoride) for Solution-Processed Ferroelectric Thin Films. Nat. Mater.2013, 12 (5), 433–438. |
| Polycaprolactone (PCL) | Hu, H.; Dorset, D. L. Crystal Structure of Poly(Iε-Caprolactone). Macromolecules1990, 23 (21), 4604–4607. |
* Fractional coordinates optimized by Schrödinger.
Using the Semicrystalline Polymer Builder Panel
After building the structure (and creating a Desmond model system), the structure should be relaxed. It is recommended that you use the MD Multistage Workflow Panel, which has two built-in protocols for semicrystalline polymers. The first, Semicrystal relaxation 1, should be used as the default. The second, Semicrystal relaxation 2, is intended for the few cases where the first fails, such as when the amorphous region is large and the crystal region is small. It should not be used as the default, as it has more degrees of freedom and so does not preserve the crystalline part of the structure as well.
If you want to select atoms from the crystalline region, there is a boolean atom-level Maestro property that you can use, called Crystal Atom (internal name b_matsci_crystal_atom). This property has the value True for crystal atoms and False for other atoms. Thus the ASL expression atom.b_matsci_crystal_atom selects all the crystal atoms.
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
Semicrystalline Polymer Builder Panel Features
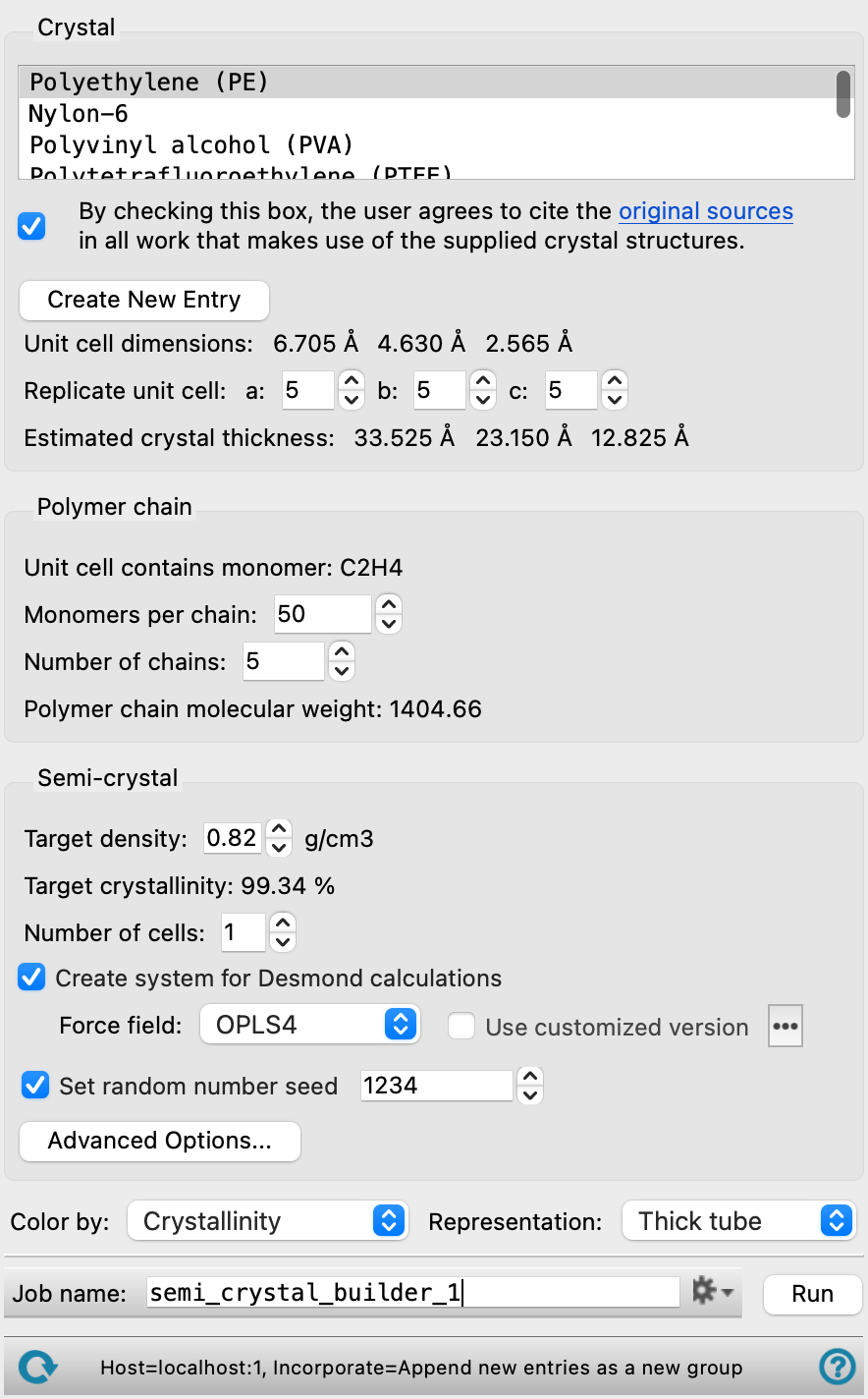
- Crystal section
-
In this section you choose the crystalline polymer you want to use, and create a slab of the desired size for the crystalline region. The monomer unit for building chains in the amorphous region is taken from the polymer you choose.
- Polymer list
-
Choose a crystalline polymer from a set of standard crystal structures, or import the crystalline polymer from the Workspace. If you import your own crystalline polymer, you should ensure that the chains are aligned along the c axis. If the structure is not orthorhombic, a new orthorhombic cell is created by default. If the input structure is triclinic, you can choose to have a triclinic cell instead of orthorhombic in the Semicrystalline Advanced Options Dialog Box. This may be important to obtain equilibration of the system.
- Citation agreement checkbox
-
You must agree to cite the original sources for the crystal you choose in all work that uses the crystal structure. This box must be checked before you can create a new entry from a supplied crystal structure (to use for building the semicrystalline structure) or run a job. The sources are listed above. It is not necessary to check this box if you supply your own crystal structure, by importing from the Workspace. The check box remains checked unless you clear it.
- Create New Entry button
-
Create a new project entry for the chosen crystal structure. The semicrystalline polymer that is built from the crystal structure is stored in this entry.
- Unit cell dimensionstext
-
This text shows the a, b, and c axis lengths of the unit cell of the chosen crystal.
- Replicate unit cell text boxes
-
These text boxes allow you to enter the number of replicas of the unit cell to create along each lattice vector, a, b, and c. This allows you to create a crystal slab of the desired size and shape.
- Estimated crystal thickness text
-
This text shows the dimensions of the crystal layer (lamella) constructed by the replication.
- Polymer chain section
-
Specify the total number of polymer chains and the length of the polymer chains in the final structure. Each chain in the final structure is of the length specified by the number of monomers per chain. You should ensure that the number and length of the chains gives an appropriate density: the density is reported in the Semi-crystal section, once the thickness of the amorphous layer is defined.
- Unit cell contains monomer text
-
This text identifies the monomer contained in the crystal by its empirical formula. This monomer unit (which may consist of several actual units) is used to build the amorphous polymer chains.
- Monomers per chain text box
-
Specify the number of monomers to use for each chain. The monomer unit is the one identified in the unit cell and reported above.
- Number of chains text box
-
Specify the number of chains to use in the final structure. The number of chains affects the target crystallinity (reported in the Target crystallinity text, below).
- Polymer chain molecular weight text
-
This text shows the molecular weight of the polymer chain.
- Semi-crystal section
-
Specify parameters for the amorphous layer.
- Target density text box
-
Specify the target density of the final cell. The target density cannot exceed the crystal density. The default is set to 70% of the crystal density.
This value is used to calculate the lamella thickness, assuming 100% crystal coverage (that is, all chains in the crystal are connected to chains in the amorphous region).
- Target crystallinity text
-
This text reports the percentage of the final structure that should be crystalline, by number of atoms, on the basis of 100% crystal coverage. The value changes with the number of chains specified in the Polymer chain section. This value is used to guide the linking of amorphous chains to crystalline chains so that the target percentage of crystalline atoms is met. The final crystallinity of the output structure is reported in the Project Table as Crystallinity.
- Number of cells text box
-
Specify the number of cells to generate for the current settings. Each cell is generated with the same settings but a different random seed. This allows you to average properties over the replicated cells, for example.
- Create system for Desmond calculations option
-
Create a model system for molecular dynamics simulations with Desmond (
.cmsfile output), which includes force field information for the simulation. If this option is selected, the force field controls are activated.- Force field option menu
-
Choose the force field for the simulations.
- Use customized version option
-
Use your customized version of the OPLS4 or OPLS5 force field, rather than the standard version in the distribution.
If the customized version is missing or invalid, the text of this option turns orange and an orange warning icon is displayed to the right, with a tooltip about the problem.
- Parameter set button
-
Select the set of custom parameters for the OPLS4 or OPLS5 force field. Opens the Set Custom Parameters Location Dialog Box.
- Set random number seed option and text box
-
Select this option to specify a random seed for all random processes used in building the polymer. Specifying the seed allows you to reproduce the results, unless other factors affect them. If this option is not selected, a seed is chosen at random.
- Advanced Options button
-
Specify options for the construction of the amorphous layer. Opens the Semicrystalline Advanced Options Dialog Box.
- Appearance settings
-
Make settings for the appearance of the output structures.
- Color by option menu
-
Color the output structure by crystallinity (the default), by molecule number, or by element. Coloring by crystallinity colors the crystalline region by element and the amorphous region magenta. The other two schemes are the standard Maestro schemes (see Coloring Atoms).
- Representation option menu
-
Choose the molecular representation for the structures, from the standard set. The default is wire.
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Semicrystalline Polymer Builder - Job Settings Dialog Box, where you can make settings for running the job.
- Status bar
-
The status bar displays information about the current job settings and status for the panel. The settings includes the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
Use the Reset button
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel. The status bar also contains the Help button
 , which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a
, which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a  button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.
button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.