Crosslink Polymers Panel
Build polymers from a box of monomers, by linking units according to a reaction scheme in which two specified bonds are broken and two new bonds are formed. Links are made when the atoms that form bonds are within a specified distance. MD at elevated temperature is used to "shake up" the mixture after each round of bond formation so that new bonds can be formed.
To open this panel: click the Tasks button and browse to Materials → Classical Mechanics → Crosslink Polymers → Crosslink Polymer Calculations .
The following licenses are required to use this panel: MS Maestro, MS CG (optional), OPLS (optional), MS Force Field Applications (optional), Desmond
- Using
- Features
- Additional Resources
Using the Crosslink Polymers Panel
The primary use for this panel is for simulating the formation of thermosetting plastic materials. The monomer units are linked according to a reaction in which two bonds are broken and two bonds formed, so that the new bonds are between the units that had the old bonds. The procedure alternates between detection of potential new bonds (using cutoffs on the interatomic distances) and their formation, and short molecular dynamics simulations at elevated temperatures to move the molecules around so that the atoms of the specified bonds can come within linking distance. The procedure is terminated when either no new bonds can be formed, or a target percentage of possible links is reached. The monomer units can be either molecular structures or coarse-grained particles.
The procedure can also be applied to the case where there are multiple reactions rather than a single reaction. In this case, the reaction chemistry is defined for each reaction, and a reaction rate is supplied to guide the formation process. When selecting a bond pair to crosslink, the reaction is chosen using a kinetic model involving a Boltzmann factor for the reaction rate and a concentration factor for the number of possible bond pairs that can crosslink. The normalized product of these two factors defines a probability for a particular reaction to occur, and the reaction is then chosen at random with the calculated probabilities. The bond pair that has the smallest interatomic distance parameter for that reaction is chosen for crosslinking. As for the case of a single reaction, the procedure is terminated when no new bonds can be formed, or a target percentage of crosslinks is reached for the reaction with the fewest initial possible crosslinks.
Before using this panel, you must make a Desmond model system containing the desired monomer units for each simulation. The Disordered System Builder Panel provides an ideal way of constructing a disordered system of molecules of a specified composition for this purpose.
Once you have the simulation box (model system), open the panel and select the entries that contain the model systems. You can then define the bonds to be broken and make threshold settings for the bonds to be formed, set up a temperature schedule for the process, specify the crosslink saturation, limit on unproductive iterations, and the parameters for the MD simulations. You can run multiple simulations in the same job, but you should ensure that the settings you make apply to all model systems. You can also compute data for detailed analysis of the crosslinking run.
You can then run the job. As Desmond is optimized for GPU execution, you should run the job on a host that has GPU cards installed and a queuing system for managing the jobs. See Preparing for Batch Queue Submission for more information.
You can run calculations on multiple systems simultaneously, by using multiple project entries as input. Each system is run as a separate job, and you can specify the number of processors or GPUs to use for each job in the Job Settings Dialog Box.
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
To visualize the results, you can use the Crosslink Polymers Viewer Panel ( click the Tasks button and browse to Materials → Classical Mechanics → Crosslink Polymers → Crosslink Polymer Results). To open this panel from the entry group for the results of a job .
.
Crosslink Polymers Panel Features
- Structure source
- Define Reactions tab
- Crosslinking tab
- Crosslinking mode options
- Target crosslink saturation box and applies to breaking bond option menu
- Maximum consecutive unproductive crosslink iterations box
- Limit number of crosslinks per iteration to option and box
- Allow crosslinks between atoms from the same original molecule option
- Maximum bond order for crosslinked bond text box
- Simulation Protocol tab
- Simulation time text box
- Time step text box
- Remove subdirectories option
- Ensemble class option menu
- Temperature options
- Pressure text box
- Attempt to converge density to
- Maximum equilibration iterations box
- Robust equilibration option
- Set random number seed option and text box
- Compute options
- Force field option menu and related controls
- Barrier potential found in N direction text
- Blocked SMARTS tab
- Job and status features
Structure source
- Use structures from option menu
-
Choose the structure source for the simulations. The structures must be Desmond model systems. Simulations on multiple structures are run concurrently.
- Project Table (n selected entries)—Use the entries that are currently selected in the Project Table or Entry List. The number of entries selected is shown on the menu item.
- Workspace (n included entries)—Use the entries that are currently included in the Workspace, treated as separate structures. The number of entries in the Workspace is shown on the menu item.
- Project Table (n selected entries)—Use the entries that are currently selected in the Project Table or Entry List. The number of entries selected is shown on the menu item.
- Open Project Table button
-
Open the Project Table panel, so you can

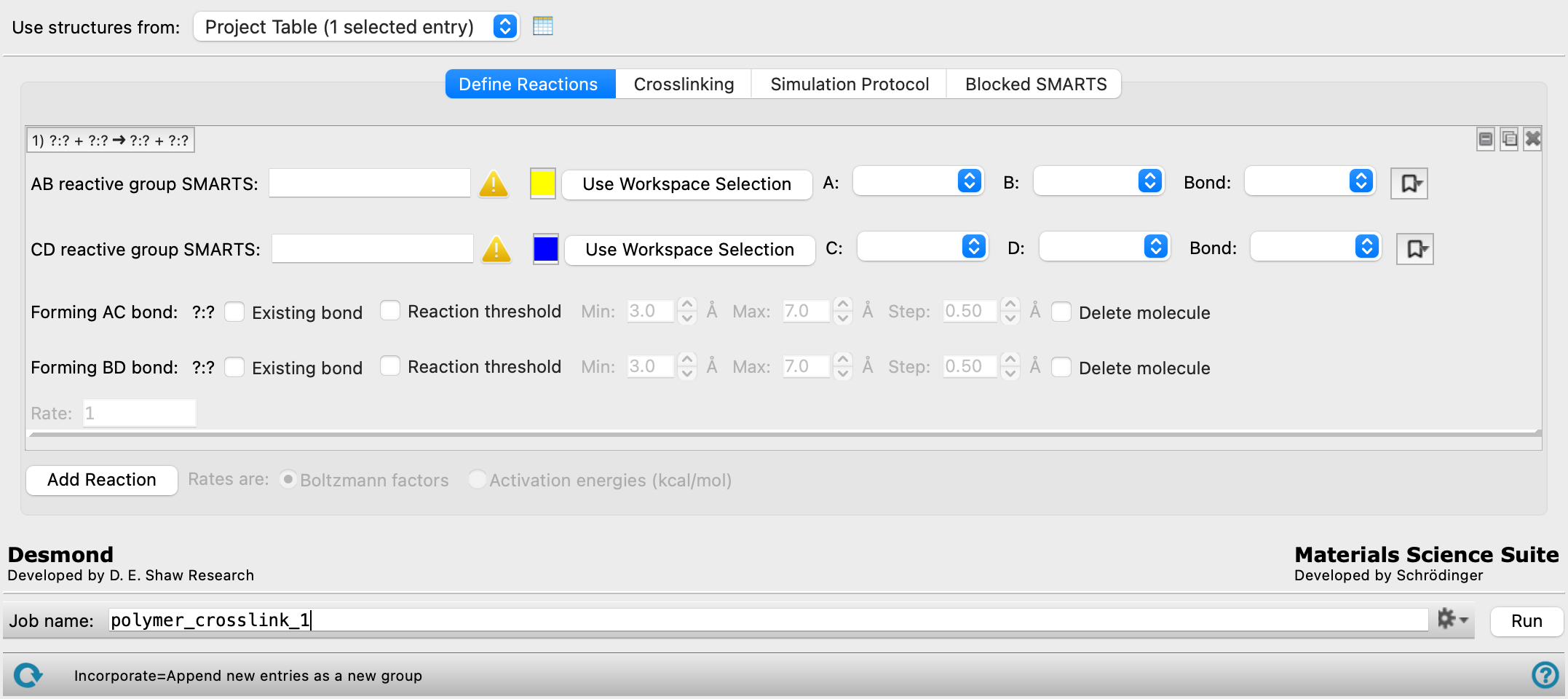
Define Reactions tab
Define one or more reactions by which the polymer is formed from the monomers. The controls for defining the reactive bonds depend on whether the monomers are molecular structures or coarse-grained particles.
For each reaction, two bonds are broken, labeled AB and CD, and exchanged to form two new bonds, labeled AC and BD. No conditions are set on where the bonds occur in the monomers: they might be on the same molecule or on different molecules. A reaction rate is also required for each reaction if multiple reactions are defined.
Each reaction has its own set of controls: a heading row, two sets of controls for the breaking bonds, two sets for the forming bonds, and a text for the reaction rate. Addition and removal of reactions is done with the Add Reaction button at the bottom of this section and the delete buttons in the set of controls for the reaction.
- Common features for reaction definition
-
These features are common to the case of molecular structures and the case of coarse-grained particles as the structure source, and so are always shown.
- Reaction label
-
The label shows the reaction in terms of the two bonds broken and formed. The label remains visible when the reaction settings are hidden.
- Reaction display and management buttons
-
These buttons allow for easy display, duplication, and deletion of a set of reaction controls.


Show or hide the reaction details. This is useful when you have a number of reactions and want to compare two separated reactions, for example. 
Duplicate the reaction. This is useful for creating a similar reaction, without having to redo all the settings. 
Delete the reaction. - Min box
-
The smallest search distance within which bonds are accepted for crosslinking. Acceptable bonds are those that would have a length shorter than or equal to the search distance.
The algorithm for finding acceptable bonds starts at the Min distance and successively increases the distance by the value in the Step box until sufficient crosslinks have been found or the value in the Max box is reached.
This box is absent if multiple reactions are defined, as the algorithm for accepting a bond only uses the maximum value.
- Max box
-
Specify the maximum search distance within which bonds are accepted for crosslinking. For a single reaction, if this value is reached and no acceptable bonds are found, the cycle is deemed unproductive, and is terminated. For multiple reactions, this distance is used when determining the number of bonds that can be formed for each reaction.
- Step box
-
Specify the increment by which the distance specified in the Min box is increased if acceptable bonding distances are not found within this distance.
This box is absent if multiple reactions are defined, as the algorithm for accepting a bond only uses the maximum value.
- Rate text box
-
Specify the rate for the reaction, either in terms of a Boltzmann factor or an activation energy in kcal/mol. The rate is used for each crosslinking event to select a reaction at random for crosslinking: the higher the rate, the higher the probability that the reaction is selected.
- Add Reaction button
-
Add a reaction to the reaction set. A new set of controls for specifying the bonds and the rate is appended to the reaction set.
- Rates are options
-
Specify whether the rates are given in terms of a Boltzmann factor or an activation energy.
- Reactive bond definitions for molecular structures
-
These settings are shown when the input structure contains normal molecular structures (i.e. composed of atoms). Definitions require the indices of atoms within a SMARTS pattern, which you can find with the SMARTS Index Panel.
- XY reactive group SMARTS text box and Use Workspace Selection button
-
Enter a SMARTS pattern for the functional group in which the bond is broken and the new bonds are formed. You can pick atoms in the Workspace and click Use Workspace Selection to define the SMARTS pattern. If the SMARTS pattern is not found in the Workspace structure (which can occur for a downstream reaction), the first two atoms in the SMARTS pattern are taken to define the reactive bond.
For a tutorial introduction to SMARTS, see http://daylight.com/dayhtml_tutorials/languages/smarts/index.html. Examples of SMARTS patterns are illustrated at http://daylight.com/dayhtml_tutorials/languages/smarts/smarts_examples.html.
- Color button
-
Color used to highlight the breaking bond in the Workspace. You can change the color by clicking the button and selecting a new color in the color selector that opens.
- Bonding atom option menus
-
Choose the atoms in the SMARTS pattern to which the new bonds will be formed. These are labeled A, B, C, and D. The bonds are formed between atoms A and C and atoms B and D. The option menus list the atoms in the SMARTS pattern as
index:element. If the SMARTS pattern does not match any atoms in the Workspace, the first atom in the SMARTS pattern is taken as atom A (or C) and the second is taken as atom B (or D). - Bond option menu
-
Choose the bond in the SMARTS pattern that is broken. The option menu lists the bonds in the SMARTS pattern, with each atom specified as
index:element.When a bond is entered, the structures are checked to see if they contain that bond, and the number of bonds is reported to the right. A green check mark indicates that the bond is valid and matches bonds in the system. The number of matching bonds can be compared to the number of molecules in the model system to verify that the SMARTS pattern is matching the desired number of bonds per molecule. The bonds are highlighted in the Workspace, with different colors for AB bonds and CD bonds.
You can choose a bond to break that does not occur in the initial structure (i.e. in the monomers). This allows a bond that has been formed by one reaction to be broken in another reaction. A warning symbol is displayed in the row for the bond and a warning is posted before running the job if the bond is not present. When the job is run, if the bond cannot be formed because there are no instances of it, the reaction is simply skipped at each step where there are no instances of the bond.
- Manage SMARTS patterns button
-
Click the bookmark icon (
 ) to open a menu of standard and previously saved SMARTS patterns. Select Save selection to save the current SMARTS pattern and bond settings. A dialog box opens, in which you can name the selection. The selection is added to the top of the menu, and is saved for future Maestro sessions in your Schrödinger user resources directory. You can delete a previously saved selection using the trash can.
) to open a menu of standard and previously saved SMARTS patterns. Select Save selection to save the current SMARTS pattern and bond settings. A dialog box opens, in which you can name the selection. The selection is added to the top of the menu, and is saved for future Maestro sessions in your Schrödinger user resources directory. You can delete a previously saved selection using the trash can. - Existing bond option
-
Check this box to require that the bond being formed is an existing bond; if it is not, it is skipped in the crosslinking. The bond formation therefore increases the bond order. This is useful for intramolecular reactions, such as elimination of CO2 from a urethane, water from a sugar, etc.
- Reaction Threshold option and text boxes
-
Select this option to apply thresholds for the interatomic distance to be accepted for the formation of a bond. Bond formation is only done if the interatomic distance is less than a given threshold. If a threshold is set for each bond, both bonds must satisfy the condition for bond formation. A range of thresholds can be set in the Min and Max boxes; the threshold is initially set to the Min value and is increased by the Step value if no acceptable bonding distances are found, repeating until acceptable distances are found or the Max value is reached. At this point the cycle is deemed unproductive, and terminated.
You must select this option for at least one of the forming bonds. If you only select one, the other bond is not checked for length and may be very long. Selecting both options ensures that both bonds are checked for a suitable length.
- Delete molecule option
-
Delete the molecule that contains the forming bond each time one is formed. This is useful for removing small product molecules (like water) from the resulting polymer. You can only do this for one of the forming bonds: selecting the option for the other bond clears the option for the first bond.
- Reactive bond definitions for coarse-grained particles
-
These features are shown when the input structure contains coarse-grained particles. There are two rows of features, one for each particle that is involved in bond formation.
- Particle X option menu
-
Choose a particle type that will be used to form bonds. The particles in the system are listed on this option menu.
- Minimum bonds option menu
-
Set the minimum number of bonds that a particle must have to be selected for crosslinking. The number of bonds a particle has can change, so this criterion is applied at each round of crosslinking.
- Maximum bonds option menu
-
Set the maximum number of bonds that a particle must have to be selected for crosslinking. The number of bonds a particle has can change, so this criterion is applied at each round of crosslinking.
- Color button
-
Color the particles of this type in the Workspace with the color on this button. Click the button to open a color selector and choose the color.
- Restrict angle to option and boxes
-
Restrict the bond angles between particles to the range specified in the text boxes.
Crosslinking tab
Specify parameters that control when the crosslinking is considered to have finished and the job stops.
- Crosslinking mode options
-
Specify the type of crosslinking calculation to run from the following options:
-
Standard—runs short MD simulations between crosslinking iterations. This allows the monomers to move after an unsuccessful crosslinking attempt, potentially coming within bonding distance during a subsequent iteration. It also allows the system to relax after a successful crosslinking event involving the formation and breaking of bonds. This mode has the longest calculation time but is expected to have the highest crosslinking coversion.
-
Fast—only runs MD simulations in iterations without successful crosslinking events. After successful crosslinking iterations, only local minimization is performed. This significantly speeds up calculation time but is expected to have a decrease in crosslinking conversion compared to the Standard mode.
-
High-Throughput—only runs MD simulations in iterations without successful crosslinking events and skips structure checks after every crosslinking event to remove ring-spears. This allows for further speedup relative to the Fast mode. However, this mode can produce poor structures with new bonds formed from crosslinking through tight rings. Final structures from this mode should be verified using the Locate Rings and Spears Panel.
-
- Target crosslink saturation box and applies to option menu
-
Specify the percentage of the available breaking bonds or particles that are required to form crosslinks before the calculation stops. The percentage is taken for the breaking bond or particle selected from the applies to option menu. By default this is the breaking bond or particle that has the fewest occurrences (SMARTS matches) in the initial structure, excluding bonds that have zero occurrences. For example, if bond A occurs 100 times and bond B occurs 50 times in the box of monomers, and the value specified is 75%, the job stops when 75% of the occurrences of bond B have been crosslinked.
The saturation is calculated as 100(1−N/Ni), where N is the current number of bonds or particles and Ni is the initial number of bonds or particles. For multiple reactions, this is computed for the reaction with the smallest Ni, as explained above. If a reaction produces more of this type of bond or particle, then the saturation could actually become negative if the production rate is greater than the depletion rate.
- Maximum consecutive unproductive crosslink iterations box
-
Specify the maximum number of consecutive iterations in which acceptable bonding distances are not found. The calculation terminates if this number of iterations is reached without forming bonds.
- Limit number of crosslinks per iteration to option and box
-
Select this option if you want to restrict the number of crosslinks formed in any iteration. If there are more crosslinks that meet the thresholds than this limit, the crosslinks are made in order of bond length (or sum of bond lengths, if both thresholds are specified).
For multiple reactions, this number is used to set the number of possible forming bonds that goes into the concentration factor used in the reaction probability. All possible forming bonds up to the specified maximum distance (Max value) are enumerated, then a single distance cutoff is applied for all reactions such that the reaction with the fewest bonds inside this cutoff has at least the number of bonds specified by this limit (or has as many as allowed by the maximum specified for the reaction).
If a temperature ramp is used, this box is not available, as the limit on the number of crosslinks is set as part of the ramp.
- Allow crosslinks between atoms from the same original molecule option
-
Allow crosslinks between atoms that start as part of the same molecule. Atoms that begin on different monomers but become part of the same molecule due to a crosslink reaction are always allowed to crosslink regardless of this option. Use this option, for instance, if the initial structure is entirely or almost entirely a single molecule.
- Maximum bond order for crosslinked bond text box
-
Specify the maximum bond order allowed for bonds that crosslink polymer chains. This can be used to prevent double bond formation, for example.
Simulation Protocol tab
Specify the MD parameters for equilibrating the system at the target temperature (in the nPT ensemble at atmospheric pressure). The simulation is run at a high temperature to allow movement of the monomers so that they can come within bonding distance, while ensuring that the system maintains its density fairly well. It also allows for relaxation of the system after breaking and forming bonds.
The equilibration is done in a number of iterations, each of which consists of a Brownie minimization step and an MD simulation step. After both steps, the density is checked for convergence. If you are using coarse-grained particles, the thermostat is automatically set to a DPD thermostat.
- Simulation time text box
-
Specify the desired simulation time in ps. The simulation time is for a single equilibration iteration.
- Time step text box
-
Specify the time step for the simulation in fs. The value is used for each equilibration iteration. Only available for constant temperature; if a ramp is used, the time step is specified as part of the ramp.
- Remove subdirectories option
-
Remove the subdirectories for the equilibration stages. These directories contain all of the trajectory data, so you will not be able to view the trajectories for these stages if you select this option. The amount of data stored in these subdirectories can be large, and removing them on completion of the job will speed up copying back the results. Selecting this option does not affect the use of the Crosslink Polymers Viewer panel.
- Ensemble class option menu
-
Choose the ensemble class for the simulation, from NPT or NVT.
- Temperature options
-
Specify a single temperature or a temperature schedule to use for the simulation. Higher temperatures will allow for more movement and closer approach of monomers, speeding up the process.
-
Constant—Enter the temperature at which the simulations are run in the text box.
-
Ramp—Use a temperature ramp for the simulations. Click Define Ramp to set up the ramp in the Define Ramp Dialog Box.
-
- Pressure text box
-
Specify the pressure, in bar, for simulations in the NPT ensemble.
- Attempt to converge density to
-
Attempt to converge the density to the specified maximum percentage change. The check is performed after each of the two steps in each equilibration iteration.
- Maximum equilibration iterations box
-
Specify the number of iterations used to equilibrate the system. The equilibration stops if the change in density is less than the density convergence threshold, or the number of iterations is exceeded. This option is not available if density convergence is turned off.
- Robust equilibration option
-
Run the robust equilibration protocol, which is as follows:
-
Attempt crosslinking and equilibration as normal.
-
If a failure occurs, decrease the time step by half and attempt the equilibration step, and repeat if it fails, with a minimum time step of 1 fs.
-
If the equilibration step still fails, remove all the crosslinks made in step 1 and attempt to form a single crosslink, selecting a different crosslink than those already tried.
-
Equilibrate, decreasing the time step if it fails, as in step 2.
-
If equilibration fails, remove that crosslink and attempt an equilibration without any crosslinks, again using decreasing time steps if necessary.
-
Whether step 5 fails or succeeds, consider the step unproductive and proceed to the next crosslinking step.
Without this option, the job fails if the equilibration fails in the first step.
-
- Set random number seed option and text box
-
Select this option to specify a random seed to be used in the simulations. Specifying the seed allows you to reproduce the results, unless other factors affect them. If this option is not selected, a seed is chosen at random.
- Compute options
-
Compute data for analysis and display.
-
Analysis data—include the analysis stage of the crosslinking job, which computes a variety of data for analysis of the crosslinking process. The analysis can be viewed in the Crosslink Polymers Viewer Panel.
-
Free volume—calculate the free volume at each iteration of the crosslinking process, with default parameters (probe radius 1.4, grid spacing 0.5). The data is added to the intermediate Maestro files that are written after each step (to
jobname_n_summary.maegz, where n is the step number). These files are returned and can be loaded into the Free Volume Analysis Viewer Panel. The final structure contains the free volume analysis run at the end of the crosslinking process, and can also be loaded into the Free Volume Analysis Viewer Panel. The free volume is available for plotting on the Time Series tab of the Crosslink Polymers Viewer Panel.
-
- Barrier potential found in N direction text
-
Displays that a barrier potential has been detected and the axis along which it is applied. Any repulsive barriers that have been defined for the system are taken into account during the simulation. This effectively renders the system non-periodic. Barriers can be defined from the Set Barrier Potential for MD Panel.
Only present when the input structure is created with the Set Barrier Potential for MD Panel.
- Force field option menu and related controls
-
Choose the force field to use for the simulations. The available force fields and the controls related to the force field depend on whether you have an all-atom structure or a coarse-grained structure.
- All-atom:
- Force field option menu
-
Choose the force field for the simulations.
- Use customized version option
-
Use your customized version of the OPLS4 or OPLS5 force field, rather than the standard version in the distribution.
If the customized version is missing or invalid, the text of this option turns orange and an orange warning icon is displayed to the right, with a tooltip about the problem.
- Parameter set button
-
Select the set of custom parameters for the OPLS4 or OPLS5 force field. Opens the Set Custom Parameters Location Dialog Box.
- Coarse-grained:
-
The force field options depend on the Location option (see below).
- Description button
-
Display a description of the chosen force field in a separate panel.
- Location options
-
Select an option for the location of the coarse-grained force field. The force fields listed on the Force field option menu depend on this choice.
- Installation—use the force fields in the installation. These are the Martini, Martini_solvation, or Martini_full force fields [15].
- Local—import a force field from your local user resources directory. These are force fields that you have saved for your own use.
Blocked SMARTS tab
Define one or more SMARTS patterns to prevent unintended chemistry from happening during the crosslinking reactions. The blocked SMARTS are not reaction specific and are applied to all crosslinking reactions specified in the Define Reactions tab. The crosslinks are searched at every iteration of the simulation, and if the blocked SMARTS is found, that iteration is rejected. This is helpful, for instance, when you want to prevent the formation of a certain ring or set of rings during the crosslinking.
This tab is not present for coarse-grained systems.
- Stage label
-
The label indicates the stage number.
- stage management buttons
-
These buttons perform display and ordering operations on the stage. They allow for easy duplication and rearrangement of stages.
Show or hide the contents of the stage. When hidden, only the stage number, label (if any) and these buttons are displayed. This is useful when you have a number of stages and want to compare two separate stages, for example. 

Move the stage up or down one place in the list.
Duplicate the stage. This is useful for creating similar stages with variations on the settings.
Delete the stage. - SMARTS text box and Use Workspace Selection button
-
Enter a SMARTS pattern for blocking unwanted chemistry. You can pick atoms in the Workspace and click Use Workspace Selection to define the SMARTS pattern.
For a tutorial introduction to SMARTS, see http://daylight.com/dayhtml_tutorials/languages/smarts/index.html. Examples of SMARTS patterns are illustrated at http://daylight.com/dayhtml_tutorials/languages/smarts/smarts_examples.html.
- Add SMARTS button
-
Add a SMARTS string to the blocked SMARTS set. A new set of controls for managing the stage is appended to the set.
Job and status features
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Crosslink Polymers - Job Settings Dialog Box, where you can make settings for running the job.
- Status bar
-
The status bar displays information about the current job settings and status for the panel. The settings includes the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
Use the Reset button
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel. The status bar also contains the Help button
 , which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a
, which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a  button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.
button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.