Complex Enumeration and Stability Analysis Panel
Enumerate complexes defined by a central atom and a specified number of ligands, and perform stability analysis on the resulting complexes. The ligands can be monodentate, bidentate, or haptic. Complexes can be composed of any combination of ligands of any type, and hence may vary in their coordination number or number of ligands.
To open this panel: click the Tasks button and browse to Materials → Enumeration → Complexes.
For a tutorial, see Organometallic Complexes.
The following licenses are required to use this panel: MS Maestro, Jaguar (optional)
- Using
- Features
- Additional Resources
Using the Complex Enumeration and Stability Analysis Panel
This panel generates a list of structures where a metal center is attached to combinations of a specified set of ligands. The coordination complexes that are enumerated can be heteroleptic or homoleptic, and multidentate ligands (e.g. κ-2 acac) and multihaptic ligands (e.g. η-5 Cp) are supported. The stability of the resulting complexes with respect to ligand exchange can optionally be evaluated with DFT.
The initially-built geometries of the complexes can be cleaned up with a force field or xTB minimization, which is often necessary to reduce steric clashes. The Stability Analysis option includes full geometry optimization with DFT. Alternatively, you might want to run a Jaguar DFT optimization on the complexes before using them for other purposes. Another way to process the results is to use the Organometallic Conformational Search Panel to generate conformers while fixing the coordination; this panel also provides an option to run a Jaguar optimization.
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
You can create single complexes with ligands of any denticity or hapticity in the Single Complex Builder Panel or the Sculpt Complex Panel.
Complex Enumeration
The ligands can be taken from up to two ligand libraries ('primary' and ‘secondary’).. Libraries can be generated in the Ligand Creator Panel, and are stored in your User resources directory. All possible combinations of ligands in the library, within the specified constraints, are taken when enumerating the complexes.
Ligands can alternatively be taken from selected entries in the Workspace or Project Table, if these entries are recognized as coordination complexes. The set of unique ligands from all the entries are used, and all possible combinations of the set, within the specified constraints, are taken when enumerating the complexes.
The titles of the complexes are assigned using the central atom symbol and the ligand name or molecular formula.
The user can specify whether the complexes generated from the enumeration should have the same number of ligands each with various coordination numbers, or the same coordination number with various numbers of ligands. For example, Al(NR2)3 is 3-coordinate at Al, whereas Al(acac)3 is 6-coordinate at Al, but both have three ligands.
Note that each ligand is treated as an equal candidate for enumeration, ignoring its charge state and the resulting oxidation state of the metal. Haptic ligands are counted as singly coordinated. The maximum coordination number is 9. When enumerating complexes with high coordination numbers, be aware that this could take considerable time, due to the large number of possible arrangements of ligands for each complex. There is also a limit of 16 bonds between the central atom and its ligands, when counting each coordinating atom of a haptic ligand separately (e.g., the cyclopentadienyl ligand is counted as having 5 bonds to the central atom).
This panel can produce redundant complexes, so it might be necessary to examine and filter the results so as to eliminate the redundant complexes. The panel provides a means to remove duplicates, which are detected by superposition; however it does not perform any rotation of the ligands around the bonds to the central atom, so some redundancies might still remain, especially for haptic ligands.
Stability Analysis
You can choose the Stability analysis with DFT option to perform a geometry optimization and calculate the ligand exchange stability for each complex generated through the enumeration. Ligand exchange stability is a way to compare the relative DFT energies within a family of heteroleptic complexes from the enumeration, indicating which combination of ligands is most energetically favored.
There is a homoleptic reference complex for every unique ligand in a heteroleptic complex. The ligand exchange stability for the heteroleptic complex is calculated as the energy required to exchange ligands of the homoleptic reference complexes with the heteroleptic complex. For example, a hetereoleptic complex of a central atom M with 2 A ligands and 1 B ligand attached, whose molecular formula can be written as MA2B, has two homoleptic reference complexes: MA3 and MB3.
The ligand exchange reaction for this example can be written as:

and the corresponding ligand exchange stability ΔELES in kcal/mol is calculated as:

where the terms of the equation are the optimized DFT energies of the heteroleptic complex and homoleptic reference complexes, respectively.
By definition, the ligand exchange stability ΔELES of a homoleptic complex is zero. A negative ligand exchange stability value means the complex is more stable with respect to its homoleptic reference complexes. The complex with the lowest ligand exchange stability within a set of enumerated complexes has the most optimal combination of ligands. It is important to note that the ligand exchange stabilities are method-dependent, and only provide approximate quantitative insights into complex stability under experimental conditions.
The equation for the ligand exchange stability for each complex in the stability analysis is explicitly reported at the end of the jobname-driver.log file. The ligand exchange stability is saved as a property in the project entry.
Complex Enumeration and Stability Analysis Panel Features
- Primary ligand source options
- Secondary ligand source option
- Libraries buttons
- Complex tab
- Isomers tab
- Job toolbar
- Status bar
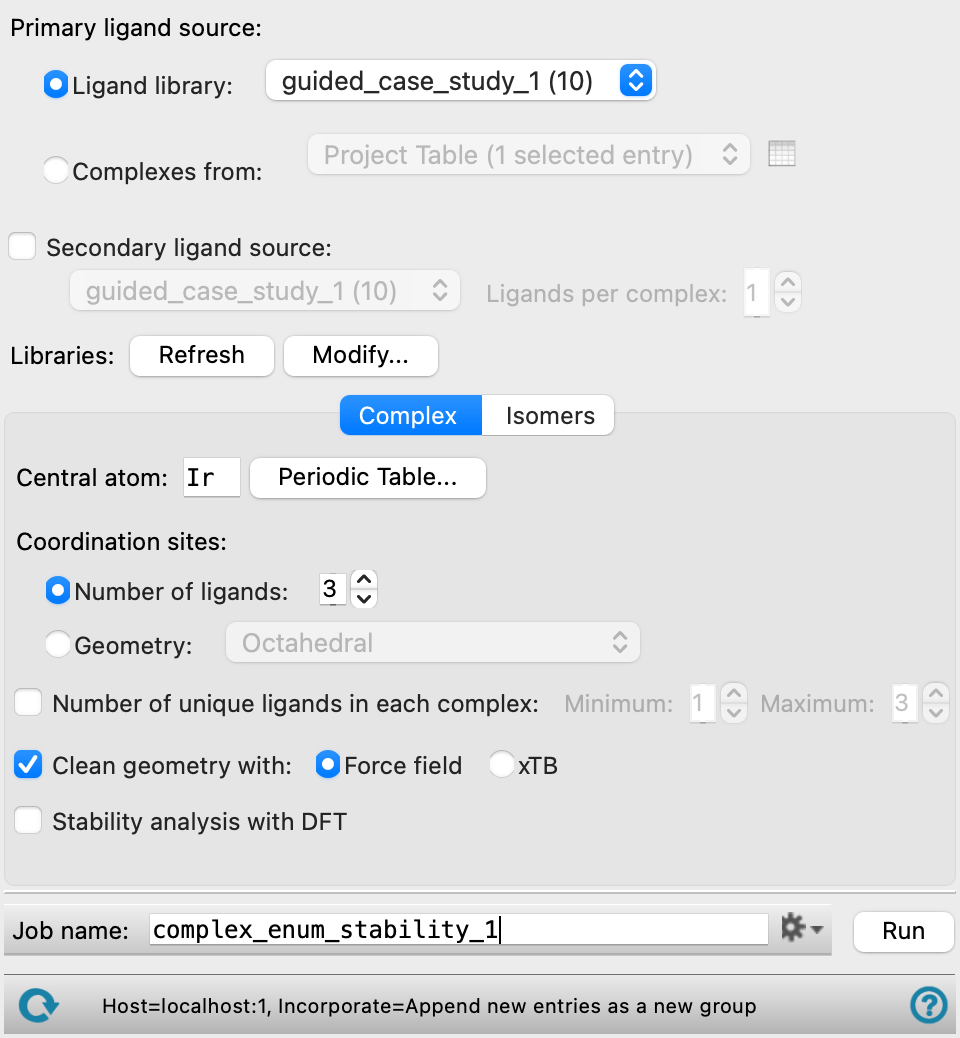
- Primary ligand source options
-
Choose the primary source of ligands for the enumeration, either directly from a Ligand Library or extracted from Complexes from project entries.
- Ligand Library
-
Choose the library for the ligands from the option menu. The number of ligands in each library is displayed in parentheses after the library name. The libraries are stored inside your User resources directory, and can be updated and managed from the Ligand Creator Panel. This menu is populated when the panel opens. If you update libraries while the panel is open, click Refresh to repopulate the menu. The latest version of the chosen library is used when you click Run, regardless of whether the menu has been refreshed.
-
Only present if Ligand Library is selected as the Primary ligand source option.
- Complexes from
-
Choose coordination complexes from which ligands will automatically be extracted as the source for ligand enumeration. Duplicate ligands will be ignored. At least one chosen entry must be the structure of a coordination complex from which a ligand can be obtained.
- Complexes from option menu
-
Choose the structure source for ligand enumeration.
- Project Table (n selected entries)—Use the entries that are currently selected in the Project Table or Entry List. The number of entries selected is shown on the menu item.
- Workspace (included entry)—Use the entry that is currently included in the Workspace. Only one entry must be included in the Workspace.
- File—Use the specified file. When this option is selected, the File name text box and Browse button are displayed.
- Project Table (n selected entries)—Use the entries that are currently selected in the Project Table or Entry List. The number of entries selected is shown on the menu item.
- Open Project Table button
-
Open the Project Table panel, so you can

- File name text box and Browse button
-
Enter the file name in this text box, or click Browse and navigate to the file. The name of the file you selected is displayed in the text box.
Only present if Complexes from is selected as the Primary ligand source option.
Optionally, choose a secondary source of ligands for the enumeration. The secondary ligand source must be a ligand library.
This can be useful, for instance, for creating complexes that all have one ligand in common.
- Library option menu
-
Choose the library for the ligands. The number of ligands in each library is displayed in parentheses after the library name. The libraries are stored inside your User resources directory, and can be updated and managed from the Ligand Creator Panel. This menu is populated when the panel opens. If you update libraries while the panel is open, click Refresh to repopulate the menu. The latest version of the chosen library is used when you click Run, regardless of whether the menu has been refreshed.
Only available if the Secondary ligand source option is selected.
- Ligands per complex text box
-
Specify the number of ligands that each complex should have from the Secondary ligand source. Enter the value in the box, or use the arrows to adjust the number of ligands.
-
Only available if the Secondary ligand source option is selected.
Manage the ligand libraries. These buttons are only available if the Library option is selected as the Primary ligand source or the Secondary ligand source option is selected.
-
Refresh—Click Refresh to repopulate the library option menus after updating the ligand libraries while the panel is open. The latest version of the chosen library is used when you click Run, regardless of whether the menu has been refreshed.
-
Modify—Modify the ligand libraries. You can add or delete ligands, and create or delete libraries. Opens the Ligand Creator Panel, where you can perform these tasks. After modifying libraries, click Refresh to update the library option menus.
Define the type of complex to enumerate.
- Central atom text box and Periodic Table button
-
Enter the element symbol for the central atom in this text box, or click the Periodic Table button to choose the central atom from a periodic table. When Workspace (included entry) is selected in the Use structures from option menu, the text box is automatically filled with the central atom from the included entry. When Project Table is selected in the Use structures from option menu, the text box is automatically filled with the central atom from the first included entry.
- Coordination sites options
-
Choose an option for how the coordination of the central atom is determined. Each haptic group occupies a single coordination site. A ligand could contain multiple haptic groups, such as 1,2 diphenylethane, and each occupies one site.
- Number of ligands text box
-
Specify the number of ligands that can be added to the central atom to form a complex. Enter the value in the box, or use the arrows to adjust the number of ligands. The coordination number of the complex can vary, depending on the types of ligands added (denticity or hapticity). The maximum coordination number of the complexes is 9. When Workspace (included entry) is selected in the Use structures from option menu, the text box is automatically filled with the number of ligands on the included entry. When Project Table is selected in the Use structures from option menu, the text box is automatically filled with the number of ligands on the first included entry.
- Geometry option menu
-
Choose the desired geometry of the ligand coordination sites around the central atom. The ligands are chosen to fill the coordination sites. The number of ligands can vary between complexes, depending on the denticity or hapticity.
- Number of unique ligands in each ligand option and Minimum/Maximum text boxes
-
Set the minimum and maximum number of unique ligands allowed in each complex.
-
To generate only homoleptic complexes, i.e. the same ligand is used at all sites, the complex formula is MLn and both the Minimum and Maximum values must be set to 1. Note that by definition, the ligand exchange stability of a homoleptic complex is zero.
- Clean geometry with option
-
Optionally, perform a minimization on the complexes and specify the method to use for the minimization.
- Force field—Minimize the geometry of the complexes with force-field minimization.
- xTB —Minimize the geometry of the complexes with GFN2-xTB. GFN2-xTB is a fast, quantum mechanics-based method and may yield more accurate geometries for many complexes at little additional computational cost.
- Stability analysis with DFT option
-
Perform geometry optimization on the enumerated complexes and compute ligand exchange stability in kcal/mol for each complex. The method for the geometry optimization can be specified by clicking the Jaguar Options button. The text below this option shows the maximum number of structures to be optimized with your selected method. The progress of the geometry optimization calculations can be viewed in the
jobname-driver.logfile. The ligand exchange stability is saved as a property in the project entry under "Ligand exchange stability (kcal/mol)". See Stability Analysis.- Options button
-
Set Jaguar options for the geometry optimization. Opens the Jaguar Options - Dialog Box. This dialog box also allows you to specify additional Jaguar keywords. The solvent (if any), level of theory, and basis set are shown to the right of the button.
- Return all Jaguar files option
-
Return the job files for all the Jaguar jobs to the job directory on the launch host. By default the files for the optimization of the individual complexes are not returned. If the job is large, copying back these files could take some time. Failed jobs are automatically copied back regardless of this setting.
Specify how isomers of various coordination numbers are treated.
- 4-coordinate geometry options
-
Select the desired geometry for 4-coordinate complexes, from Tetrahedral or Square planar. Only available when relevant for the complex.
- Bidentate ligands options
- Flip orientation to create isomers option
-
Enumerate all possible isomers of complexes with asymmetric bidentate ligands by including both possible orientations of these ligands.
- Do not flip option
-
Select the desired isomer to generate for square planar or octahedral isomers.
Remove duplicate complexes from the enumerated set. The duplicates are detected by superimposing complexes of the same formula and testing the distances between corresponding atoms. If all atoms in a complex are within a defined tolerance of the corresponding atoms in a reference complex, the complex is considered to be a duplicate of the reference, and is removed.
The removal of duplicates is slower than the enumeration, by a factor of about 5 or more.
- Count optical isomers as duplicates option
-
Remove optical isomers as well as identical complexes. Optical isomers are generated by inversion of the coordinates around the central atom atom.
- Tolerance text box
-
Set the tolerance for detecting atoms that are considered to be at the same location. Any atom in a complex that is closer than this amount to the corresponding atom in another complex is considered to be at the same location. The default tolerance might not eliminate redundant haptic ligands that differ only by the rotation about the bond between the ligand and the central atom. A larger threshold can eliminate this kind of duplicate, but it should not be made too large (larger than 2 Å) or it may eliminate complexes that are not duplicates.
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Complex Enumeration and Stability Analysis - Job Settings Dialog Box, where you can make settings for running the job.
The status bar displays information about the current job settings and status for the panel. The settings includes the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
Use the Reset button  to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel.
The status bar also contains the Help button  , which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a
, which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a  button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.
button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.