Crystal Structure Prediction Panel
Predict possible crystal polymorphs and rank them by their energy.
To display this panel
Note
Please note that Job Server is required for submission of crystal structure prediction jobs. To learn more about Job Server, see Job Submission to a Cluster.
The following licenses are required to use this panel: Crystal Structure Prediction, Force Field Builder, Quantum Espresso Interface, Jaguar, MacroModel, Desmond
- Using
- Features
- Additional Resources
Using the Crystal Structure Prediction Panel
The Crystal Structure Prediction Panel can be used to explore potential crystal polymorphs and rank them based on their energies. This panel provides insight into the stability of polymorphs without the expense and time-intensiveness of experimental polymorph screening. The input to the panel is a single molecule or a group of conformers previously generated using this workflow. The crystal structure prediction workflow consists of the following steps:
-
Conformer Generation: Conformers are generated for the input molecule. Related files can be found in the job directory under
output_confgen.maegz. It is recommended to run only this step on a molecule first to ensure the conformers are what one would expect before moving to the remainder of the workflow. -
Packing Search: Pack the conformers into unit cells using the space groups displayed in the Advanced Settings dialog box. Related files can be found in the job directory under
output_packing_search.maegz. -
Molecular Dynamics (MD) Relaxation: A molecular dynamics simulation is run on each crystal structure generated by the packing search. The outputs with high energy, >10 kcal/mol relative to the lowest energy structure, are removed prior to the next stage of ranking. Related files can be found in the job directory under
output_md_relaxation.maegz. -
Machine Learning Force Field (MLFF) Relaxation: The crystal structures are then relaxed using MLFF and the outputs with high energy, >6 kcal/mol relative to the lowest energy structure, are removed. Related files can be found in the job directory under
output_qrnn_relaxation.maegz. -
Density Functional Theory (DFT) Relaxation: Relax the crystal structures using DFT via Quantum ESPRESSO. The crystal structures are first relaxed with the PBE density functional with loose thresholds. Followed by single point energy calculations using the r2SCAN density functional. Next, the structures are relaxed using the PBE functional with tight thresholds. Finally, single point energy calculations using the r2SCAN density functional are performed. In all of the calculations, DFT-D3 dispersion is used. Crystal structures with high energies are removed after each step. Please note that calculation time increases significantly for larger structures. Related files can be found in the job directory under
output_dft_relaxation.maegz.
To visualize the results, you can use the Crystal Structure Prediction Viewer Panel (click the Tasks button and browse to Polymorph Prediction → Crystal Structure Prediction Analysis).
Please see the reference [87] to learn more about this crystal structure prediction workflow.
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
Crystal Structure Prediction Panel Features
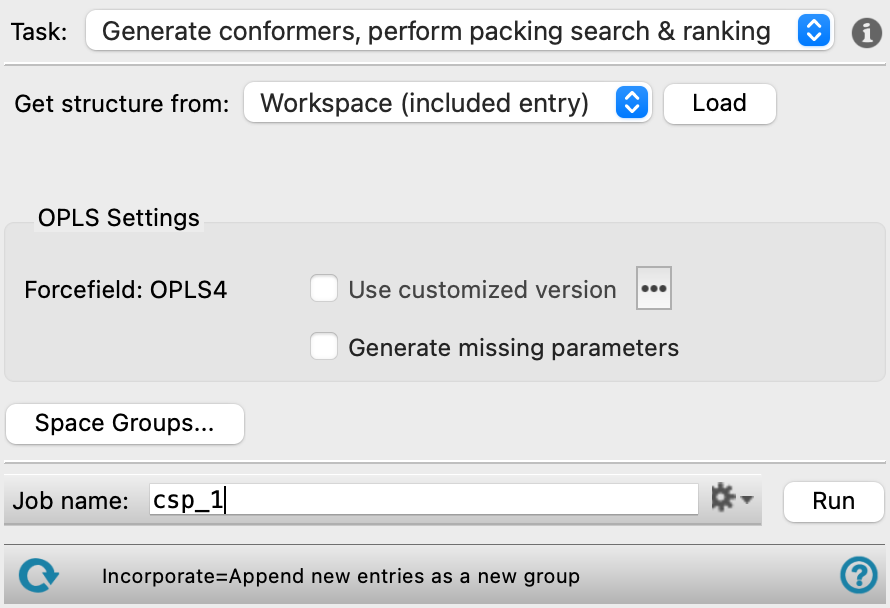
- Task option menu
-
Choose the crystal structure prediction calculation type to run from the following options:
- Generate conformers, perform packing search & ranking—Generate conformers of the input molecule, pack the conformers into unit cells using the space groups from the Advanced Settings dialog box, relax the crystal structures, and filter them based on their energies.
- Generate conformers only—Generate conformers of the input molecule. It is recommended to run this step on a molecule first to ensure the conformers are what one would expect before moving to the significantly longer packing and ranking calculation.
- Perform packing search & ranking—Pack the conformers into unit cells using various space groups, the crystal structures are relaxed, and filtered based on their energies. Load conformers previously generated using the Generate conformers only task type using the Workflow Action Menu (
 ).
).
See the Using the Crystal Structure Prediction Panel for more.
- Structure information button
-
Click on the information icon on the top right of the panel to display the input required for the task type. A single molecule is the required input for the Generate conformers, perform packing search & ranking and Generate conformers only tasks. A group of conformers calculated using the Generate conformers only task type is the required input for Perform packing search & ranking.
- Get structure from option menu
-
Choose the structure source for the single molecule or group of conformers.
- Project Table (selected entries)—Use the entries that are currently selected in the Project Table.
- Workspace (included entry)—Use the entry that is currently included in the Workspace. Only one entry must be included in the Workspace.
- File—Use the specified file. When this option is selected, the File name text box and Browse button are displayed.
-
- Load button
-
Load the molecule or conformer group from the
- Browse button
-
Click Browse and navigate to the desired file
- Loaded structure text
-
Displays the name of the loaded molecule or the number of conformers loaded. For molecules, a green check-mark icon is displayed to the right of the molecule name. If the input molecule has torsion angles with missing force field terms, the Generate missing parameters option is selected by default. Unselecting this option will result in a stop sign icon being displayed to the right of the molecule name. Select the Generate missing parameters or Use customized version option to continue. Noneditable.
- OPLS Settings section
-
- Force field task
-
Displays the force field for the crystal structure prediction calculation. The OPLS4 force field is used.
- Use customized version option
-
Use your customized version of the OPLS4 force field, rather than the standard version in the distribution.
If the customized version is missing or invalid, the text of this option turns orange and an orange warning icon is displayed to the right, with a tooltip about the problem.
- Parameter set button
-
Select the set of custom parameters for the OPLS4 force field. Opens the Set Custom Parameters Location Dialog Box.
- Generate missing parameters option
-
Run quantum mechanical calculations on the molecule and fit the results to generate any missing force-field torsional parameters before running the crystal structure prediction job. Only available for the Generate conformers, perform packing search & ranking and Generate conformers only tasks.
- Space Groups button
-
Opens the Advanced Settings dialog box, which lists the space groups available to use. Restrict packing search to the space groups selected in the dialog. Click Select All to use all the listed space groups. Click Save to confirm choices of space groups and exit the pane. Click Cancel to exit the pane without saving any changes.
-
For chiral molecules, by default only Sohncke groups are selected so that the predictive crystal structures are enantiomer pure crystals.
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Crystal Structure Prediction - Job Settings Dialog Box, where you can make settings for running the job.
Job Server is required for submission of crystal structure prediction jobs. To learn more about Job Server, see Job Submission to a Cluster.
- Status bar
-
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel.If you can submit a job from the panel, the status bar displays information about the current job settings and status for the panel. The settings include the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
The status bar also contains the Help button
 , which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.
, which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.