Adsorption Energy Calculations Panel
Calculate the adsorption/desorption energies of molecules on substrates at 0 K and 1 atm, and the adsorption free energy at a range of temperatures and pressures, with structures from the Enumerate Adsorbates Panel, Desorption Enumeration Panel, or Adsorption Site Finder Panel.
To open this panel: click the Tasks button and browse to Materials → Quantum Mechanics → Workflows → Adsorption Energy.
To open this panel from the entry group for the results of a Enumerate Adsorbates, Desorption Enumeration, or Adsorption Site Finder job .
.
The following licenses are required to use this panel: MS Maestro, Quantum Espresso Interface, Jaguar, MS Surface (optional), MS Force Field Applications (optional)
- Overview
- Using
- Features
- Additional Resources
Overview of Adsorption Energy Calculations
The adsorption of a gas molecule B onto a solid substrate A may be written as a reaction
A(s) + B(g) → A-B(s)
To properly obtain Gibbs free energies of adsorption  , DFT phonon calculations of surface slabs can be carried out, using the Quantum ESPRESSO Calculations Panel. Instead, this panel uses a faster method to approximate these free energy differences from DFT vibrational analysis of the gas molecule only, as explained here.
, DFT phonon calculations of surface slabs can be carried out, using the Quantum ESPRESSO Calculations Panel. Instead, this panel uses a faster method to approximate these free energy differences from DFT vibrational analysis of the gas molecule only, as explained here.
Let 

where the free energy correction  depends on both temperature and pressure via enthalpic and entropic terms, see Thermochemical Properties from Jaguar Calculations.
depends on both temperature and pressure via enthalpic and entropic terms, see Thermochemical Properties from Jaguar Calculations.
In the absence of phonon calculations of zero point energy (ZPE), we set  , which results in an unquantified offset in
, which results in an unquantified offset in  at all temperatures and pressures.
at all temperatures and pressures.
Calculating  and
and  would also require phonon calculations, and in their absence we neglect any changes in vibrational motion at the surface in terms of its effect on the Gibbs free energy, i.e.
would also require phonon calculations, and in their absence we neglect any changes in vibrational motion at the surface in terms of its effect on the Gibbs free energy, i.e.  .
.
Hence 
and 
Within this approximation,  is obtained by DFT vibrational analysis with Jaguar. Since free energy is a macroscopic quantity, it does not matter that
is obtained by DFT vibrational analysis with Jaguar. Since free energy is a macroscopic quantity, it does not matter that  is calculated using a different electronic structure method than that for
is calculated using a different electronic structure method than that for  .
.
Physically, this approximation recognizes that the entropy change during adsorption is often dominated by the loss of translational and rotational entropy when gas B is immobilized on the surface.
Depending on the strength of adsorption, the adsorbate may be fixed (losing 100% of its entropy) or it may vibrate, rotate or diffuse across the surface. For the latter case, some published studies show around one third of a molecule's entropy being lost on adsorption, consistent with motion in two dimensions rather than three [42, 43, 44]. The panel therefore allows you to specify the percentage 'x' of the molecule's free energy that is lost on adsorption:  .
.
Using the Adsorption Energy Calculations Panel
Adsorption energy calculations can be run on one or more output structures from the Enumerate Adsorbates Panel, Desorption Enumeration Panel, or Adsorption Site Finder Panel. Adsorption energies are reported from structures The output of this panel is adsorption energies when the input is from the Enumerate Adsorbates Panel and desorption energies when the input is from the Desorption Enumeration Panel. Desorption energies here are the negative of adsorption energies.
The Adsorption Energy panel allows you to set a range of temperatures and pressures for the calculation of the adsorption free energy. For each combination of temperature and pressure, the values of ΔG, ΔE are returned as entry properties. These values are written to a CSV file, jobname-properties.csv.
If you want to calculate free energies of adsorption for other temperatures and pressures after running an initial set, you can use the output structures from a previous adsorption energy run.
- Make sure that only the adsorbate structures from the previous run are selected as input.
- Select Add data to completed entries.
- Set the temperature and pressure ranges, and the free energy loss (which should be the same as for the initial set for consistency).
- Click Append Free Energies to Entries to calculate the free energies and add them to the existing entries as properties.
You can view plots of energies, free energies, and entropies vs pressure or temperature in the Thermochemistry Viewer Panel. To open this panel from the entry group for the results of a job.
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
Adsorption Energy Calculations Panel Features
- Input options
- Adsorption/Desorption energy at 0K and 1 atm section
- Adsorption/Desorption free energy option and section
- Also produce energies in kJ/mol option
- Options button
- Job toolbar
- Status bar
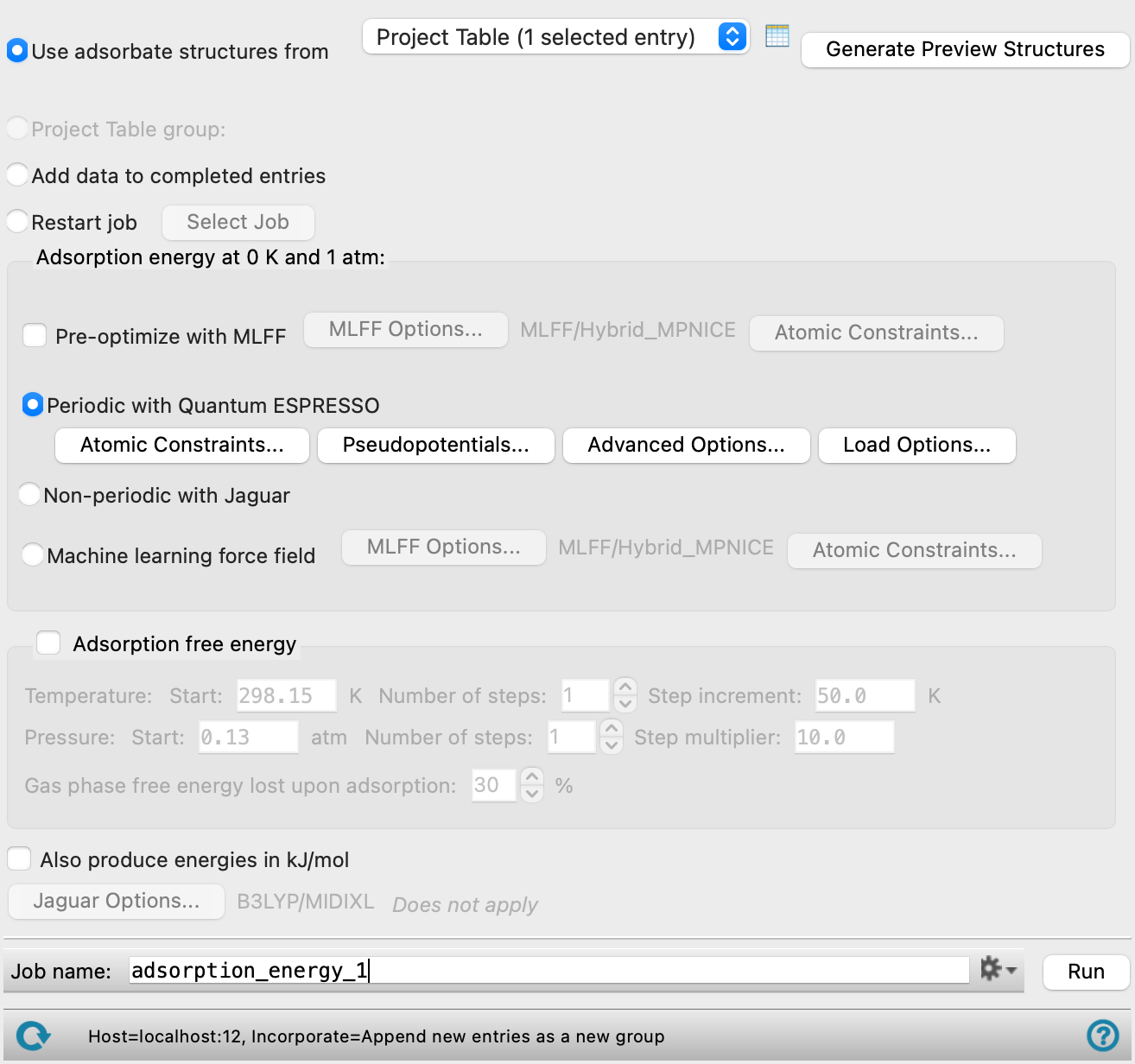
- Input options
-
Select an option for the source of the adsorbate structures.
- Use adsorbate structures from option menu
-
Choose the structure source for the substrates with adsorbates.
- Project Table (n selected entries)—Use the entries that are currently selected in the Project Table or Entry List. The number of entries selected is shown on the menu item.
- Workspace (n included entries)—Use the entries that are currently included in the Workspace, treated as separate structures. The number of entries in the Workspace is shown on the menu item.
- File—Use the specified file. When this option is selected, the File name text box and Browse button are displayed.
- Project Table (n selected entries)—Use the entries that are currently selected in the Project Table or Entry List. The number of entries selected is shown on the menu item.
-
The adsorbate structures can come from the Enumerate Adsorbates Panel or Desorption Enumeration Panel, if the optimization and calculation of adsorption/desorption energies are required, or from this panel, to add data for temperatures and pressures to the structures from a previous run. This particular input format for adsorbates allows the associated gas molecules and substrates to be constructed.
- Open Project Table button
-
Open the Project Table panel, so you can

- File name text box and Browse button
-
Enter the file name in this text box, or click Browse and navigate to the file. The name of the file you selected is displayed in the text box.
- Generate Preview Structures button
-
Generate all the initial structures for the job and add them to the project as a new entry group. This allows examination of the structures in the Workspace to fix any geometric issues before the job is run. You should be careful to change only the atomic positions and geometry of the entries (e.g. to relieve clashes).
If you click this button, the input selection option switches to Project Table group and the group name is listed to the right of the option. If you click the button multiple times, a new group is created each time you click, and the old group is renamed.
- Project Table group option
-
Use the structures in the selected project table group. This option is automatically selected when you click Generate Preview Structures, and is unavailable otherwise. The name of the group that contains the generated structures is shown to the right of the option.
- Add data to completed entries option
-
Add free energies to entries for completed jobs at additional temperatures and pressures. When you select this option, the structure source selection tools (like those shown above for Use adsorbate structures from) are displayed to the right, and you can use them to select project entries or files.
- Restart job option
-
Restart an incomplete job. When you select this option, the Locate Adsorption Energy Job dialog box opens automatically, where you can select a recently run job, as listed in the job database, or you can locate the job files by entering the path to the job directory or browsing to it. If this option is already selected, you can click the Select Job button to open the dialog box again and choose a different job.
-
When you have selected a job, the current structures of adsorbate, gas and substrate from that job are imported into the project as an entry group. You can display the structures in the Workspace to fix the geometry for the jobs that need restarting. Make sure you do not change anything other than the geometry.
-
The entry titles are shown as a list in the panel with a checkbox next to each title. The checked entries are the ones that are re-run when the job is started; the results for the unchecked entries are simply copied. The checkboxes are automatically ticked for structures that failed in the previous job, and the text (failed) is shown after the title.
You must ensure that the panel options are set to be compatible with the job that is being restarted— i.e. the same MLFF or QM job type and options should be selected as before. Alternatively, you can use this Restart option to take existing structures and run them with a different job type or job options; in this case, make sure to manually tick all of the entries so that they are all computed with the same method.
- Adsorption/Desorption energy at 0K and 1 atm section
-
Specify methods for optimizing the substrate, the gas molecule, and the "adsorbate" (here used to refer to the adsorbate+substrate system). The atomic coordinates are optimized, and the adsorption energy at 0K and 1atm is evaluated from the optimized structures. This is the time-consuming part of the calculation, and can be distributed across multiple processors. If Adsorption/Desorption free energy is also selected, frequency calculations are run on the gas molecules with Jaguar, for use in calculating the free energies.
- Pre-optimize with MLFF option
-
Optimize the geometries using a machine learning force field prior to a quantum mechanics calculations. Select this option for periodic or non-periodic systems. See Machine Learning Force Fields for more information.
You can choose settings for the MLFF calculations using the button to the right.
Only available when the Periodic with Quantum Espresso or Non-periodic with Jaguar option is selected. When Periodic with Quantum Espresso is selected, the same atomic constraints are applied to the pre-optimization and optimization and only need to be selected once. No atomic constraints are applied for optimization with the Non-periodic with Jaguar option.
- MLFF Options button
-
Set MLFF options for the geometry optimizations. Opens the MLFF Options - Adsorption Energy Dialog Box. This dialog box also allows you to specify additional Jaguar keywords. The selected model is shown to the right of the button. For systems far from optimum geometry, a high maximum number of optimization steps and “Loose” convergence criteria are recommended.
- Atomic Constraints button
-
Set or remove Cartesian constraints for atoms in the system. Opens the Atomic Constraints Dialog Box, where you can choose the constraint type (X, Y, Z, or all three), and pick atoms in the Workspace or use the Workspace selection to apply the constraints to. You can delete selected or all constraints.
- Periodic with Quantum Espresso option
-
Run the calculations using periodic boundary conditions, with Quantum Espresso. The atomic coordinates are optimized with the cell parameters held fixed. Select this option if the substrate is a slab or other periodic system such as a nanotube.
You can choose settings for the QE calculations using the buttons below. Not all the options are available in the Quantum ESPRESSO Calculations - Advanced Options Dialog Box, as the slab must remain the same as in the original slab calculation.
- Atomic Constraints button
-
Set or remove Cartesian constraints for atoms in the system. Opens the Atomic Constraints Dialog Box, where you can choose the constraint type (X, Y, Z, or all three), and pick atoms in the Workspace or use the Workspace selection to apply the constraints to. You can delete selected or all constraints.
- Pseudopotentials button
-
Select pseudopotentials for use in the calculations. Opens the Quantum ESPRESSO Calculations - Pseudopotentials Dialog Box. The set of recommended PBE ultrasoft pseudopotentials is distributed with the suite and available from the dialog box. See Installing and Configuring Quantum ESPRESSO for instructions on downloading other pseudopotential sets.
- Advanced Options button
-
Set options for the calculation: spin treatment, density functional, dispersion corrections, Brillouin zone partitioning, occupation, SCF and optimization accuracy
- Load Options button
-
Load option settings from a Quantum ESPRESSO config file (
.cfg). Opens a file selector so you can navigate to and select the config file. The settings replace those in the Quantum ESPRESSO Calculations - Advanced Options Dialog Box.
- Non-periodic with Jaguar option
-
Run the calculations as an isolated (non-periodic) system, with Jaguar. Select this option if the substrate is a nanoparticle.
- Machine learning force field option
-
Run the calculations using a machine learning force field in place of the quantum mechanics calculations. Select this option for periodic or non-periodic systems. See Machine Learning Force Fields for more information.
You can choose settings for the MLFF calculations using the button below.
- MLFF Options button
-
Set MLFF options for the adsorption energy calculations. Opens the MLFF Options - Adsorption Energy Dialog Box. This dialog box also allows you to specify additional Jaguar keywords. The selected model is shown to the right of the button. For systems far from optimum geometry, a high maximum number of optimization steps and “Loose” convergence criteria are recommended.
- Atomic Constraints button
-
Set or remove Cartesian constraints for atoms in the system. Opens the Atomic Constraints Dialog Box, where you can choose the constraint type (X, Y, Z, or all three), and pick atoms in the Workspace or use the Workspace selection to apply the constraints to. You can delete selected or all constraints.
- Adsorption/Desorption free energy option and section
-
Calculate the adsorption/desorption free energy as a function of temperature and pressure. This is very quick, as it uses the information generated in the first stage.
- Temperature text boxes
-
Specify the starting temperature in K, the number of temperature steps, and the increment in the temperature for each step.
- Pressure text boxes
-
Specify the starting pressure in atm, the number of pressure steps, and the factor by which the previous pressure is multiplied at each step.
- Gas phase free energy lost upon adsorption text box
-
Specify the percentage of the gas-phase free energy that is lost when the molecule is adsorbed. The default is in the range of values found in the literature (see Using section).
- Append Free Energies to Entries button
-
Calculate the free energies and append the energy properties to the input structures. This can only be done if the input is a set of structures from a previous adsorption energy run, as this run is the source of the information required for the free energy calculations. The new thermodynamic data is written to a new CSV file, with a name derived from the CSV file name for the original run. The Thermochemistry Viewer Panel reads the data from all the CSV files when you load the results for a run.
This button is only present if Add data to completed entries is selected. The Jaguar options button and job options are not available, as the new values are added immediately to the CSV file and the Project Table with this button, without the need for running a job.
- Also produce energies in kJ/mol option
-
Calculate the energies in kJ/mol in addition to kcal/mol. The energies in kJ/mol are added to the output structures as properties. This is very quick, as it uses the information generated in the first stage of the calculation.
- Options button
-
Set Jaguar options for the adsorption energy calculations. Opens the Jaguar Options - Adsorption Energy Calculations Dialog Box. This dialog box also allows you to specify additional Jaguar keywords. The solvent (if any), level of theory, and basis set are shown to the right of the button.
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Adsorption Energy Calculations - Job Settings Dialog Box, where you can make settings for running the job.
- Status bar
-
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel.If you can submit a job from the panel, the status bar displays information about the current job settings and status for the panel. The settings include the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
The status bar also contains the Help button
 , which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.
, which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.