Enumerate Adsorbates Panel
Build chemically reasonable initial adsorbate geometries for all combinations of a set of molecules on a set of surfaces.
To open this panel: click the Tasks button and browse to Materials → Enumeration → Adsorption.
The following licenses are required to use this panel: MS Maestro, MS Surface (optional)
- Using
- Features
- Additional Resources
Using the Enumerate Adsorbates Panel
The purpose of this panel is to build adsorbate structures for when a gas-phase molecule adsorbs on a solid substrate. The enumeration generates one or more adsorbate systems for each substrate and each gas molecule. These can be used as initial geometries for further calculations, such as locating adsorption sites with the Adsorption Site Finder Panel, periodic DFT calculations with Quantum ESPRESSO, or calculation of adsorption energies with the Adsorption Energy Calculations Panel.
Both the gas molecule(s) and the substrate(s) must exist as entries in the project first, having been previously imported, built or computed. You can then select one or more substrates in the Project Table (or include them in the Workspace), and load them into the workflow, then select or include one or more gas molecules and load them into the workflow. The panel does not allow addition or deletion of individual structures, so the selection must be done in the Project Table.
Substrates can be periodic systems such as surfaces from the Build Slabs and Interfaces Panel or nonperiodic molecules, clusters, or nanoparticles from the Nanostructure Builder Panel. The type of output structure (periodic or nonperiodic) is determined by the substrate.
A reactive atom is specified for each substrate and adsorbate molecule. The reactive atom is chosen automatically by default, as the most exposed non-hydrogen atom for the substrate and the most exposed atom for the adsorbate molecule. If two atoms are equally exposed (which is likely in a flat supercell), the one closer to the center of the cell is chosen. Using the filter, the atoms considered for the reactive atom can be restricted to a specified type of atom. Alternatively, you can pick the reactive atoms yourself in the Workspace. Other adsorption sites on the substrate in addition to the reactive atom can be selected using the Include surface atoms within option. If there are multiple substrate atoms, you can choose to adsorb at bridge and/or hollow sites, as well as on-top. Rotation of the gas molecule is also possible. Once the adsorbate structure is built, zero order bonds show where new bonding has occurred. The output of the enumeration is incorporated into the project as an entry group.
When the adsorbate structures are enumerated, there may be overlapping atoms. The structures that contain overlapping atoms are placed in a subgroup in the output, named Bad Contacts, and the overlapping atoms are highlighted in the structures. You should examine the geometries carefully before running calculations on the structures. You may need to adjust them by hand, change the reactive atoms, or increase the vacuum buffer before submitting them to QE or Jaguar. The bad contacts are defined in the same way as in the Interactions Toolbox, and the definition can be changed under Pi-Pi and Contact Criteria settings in the Preferences Panel.
To write out the input file and a script for running the job from the command line, click the arrow next to the Settings button  and choose Write.
and choose Write.
Enumerate Adsorbates Panel Features
- Substrates section
- Gas molecules section
- Adsorption types options
- Neighbor factor text box
- Adsorption direction options
- Enumerate N rotations around the adsorption axis option and text box
- Create additional pre-adsorption structure option
- Remove symmetry equivalent structures option
- Job toolbar
- Status bar
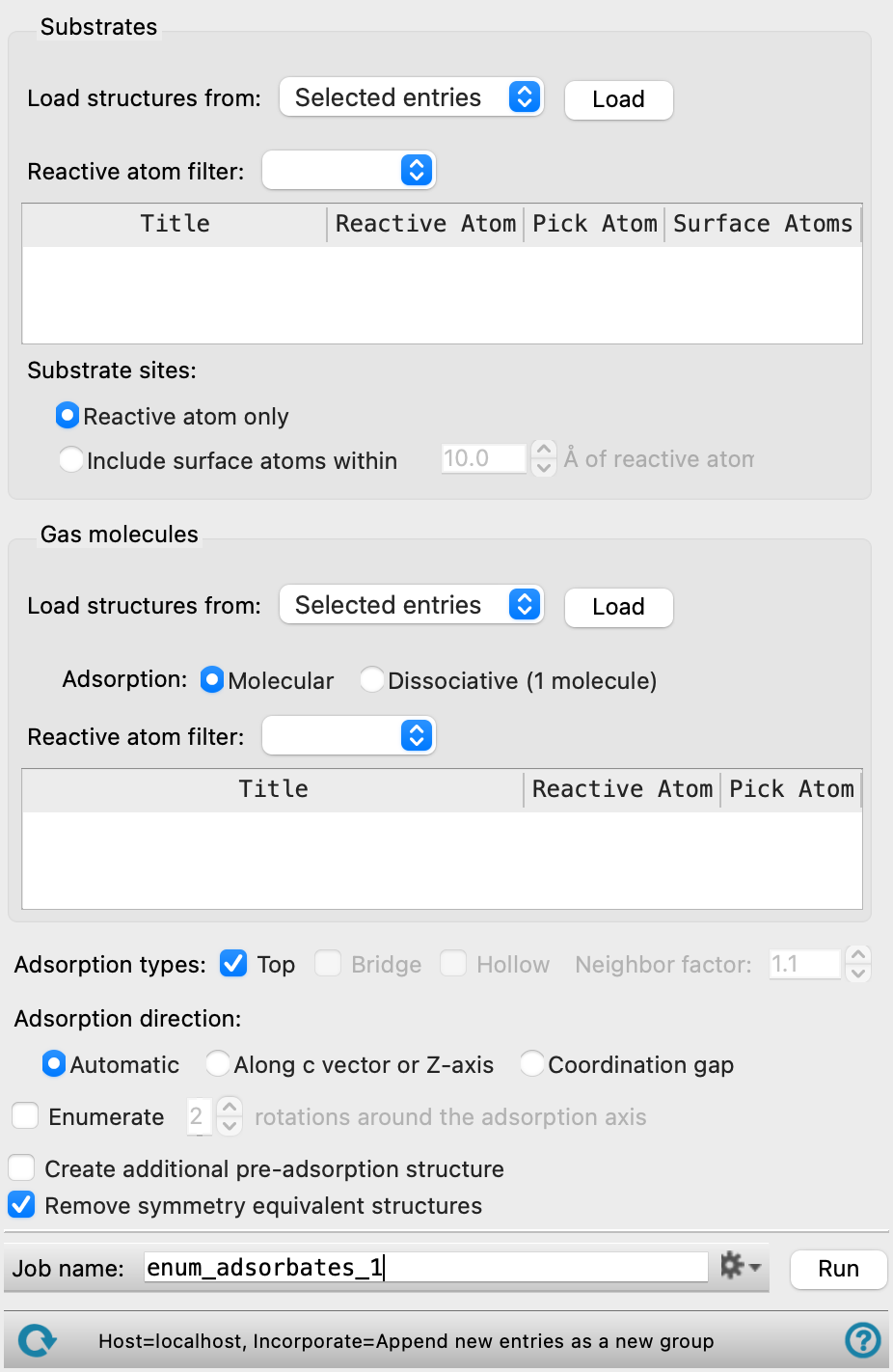
- Substrates section
-
In this section you load the substrates and specify the reactive atom for each substrate. Multiple reactive atoms can be selected for each substrate. The substrate can be a periodic structure, such as a slab prepared with the Build Slabs and Interfaces Panel, or a discrete structure such as a nanoparticle, built with the Nanoparticle Builder Panel. If the substrate is periodic, you must make sure there is enough space above the surface to accommodate the adsorbate. You can do this by specifying the vacuum buffer in the Build Slabs and Interfaces Panel, for example, or using the Redefine Lattice Panel.
- Load structures from option menu and Load button
-
Select the source for the structures, from Selected entries or Included entries. Click the Load button to load these structures into the workflow. The structures replace any existing structures.
- Reactive atom filter option menu
-
Select classes of atoms to use as a filter for the automatic selection of reactive atoms. The menu items include Heavy atoms, Any, Metals, Non-metals, the elements in the structure, SMARTS, for more complex restrictions, and Atom index. The default is Heavy atoms, meaning any non-hydrogen atom. If you choose SMARTS or Atom index, a text box is displayed for entering the SMARTS pattern/ index value.
- Structure table
-
The structures are listed in this table, identified by title. For each, the reactive atom is identified by element and atom number in the Reactive Atom column. By default, the column is automatically populated based on the selection in the Reactive atom filter option menu. You can manually pick the reactive atom from the Workspace by clicking the Pick in WS button in the Pick Atom column, and picking an atom in the structure in the Workspace. The Surface Atoms column displays how many atoms are considered an adsorption site for each structure, and updates if the Substrate sites options are changed.
- Substrate sites options
-
Specify what atoms can be considered an adsorption site. Any adsorption site is highlighted in the Workspace.
- Reactive atom only option
-
Only the atom listed in the Reactive Atom column of the Structure table is considered an adsorption site.
- Include surface atoms within option and text box
-
Atoms matching the Reactive atom filter within a specified radius of the reactive atom are also considered as potential adsorption sites. Specify the radius in angstroms in the text box. Only atoms with an exposed surface area that is at least 1% of its total surface area and on the same side of the substrate as the reactive atom are eligible. The number of atoms considered as adsorption sites for each structure is updated in the Surface Atoms column of the Structure table.
- Gas molecules section
-
In this section you load the gas molecules to adsorb to the substrate surface and specify the reactive atom for each molecule. There are no restrictions on the molecule; if it has periodic boundary conditions these are ignored.
- Load structures from option menu and Load button
-
For the Molecular option, select the source for the structures, from Selected entries or Included entries. For the Dissociative option, select the source for the structures, from Selected entry or Included entry. Click the Load button to load the(se) structure(s) into the workflow. The structures replace any existing structures.
- Adsorption options
-
- Molecular option
-
Select this option to perform molecular adsorption with the specified gas molecule(s). Multiple gas molecules can be specified for enumeration.
- Dissociative option
-
Select this option to perform dissociative adsorption with the specified gas molecule. Only one molecule can be specified for dissociative adsorption. The bonds for the molecule are broken around the selected reactive atom. Adsorption configurations are enumerated from the dissociated molecule.
- Reactive atom filter option menu
-
Select classes of atoms to use as a filter for the automatic selection of reactive atoms. The menu items include Any, Heavy atoms, Metals, Non-metals, the elements in the structure, SMARTS, for more complex restrictions, and Atom index. The default is Any, meaning no restriction on the atom chosen. If you choose SMARTS or Atom index, a text box is displayed for entering the SMARTS pattern/ index value.
- Structure table
-
The structures are listed in this table, identified by title. For each, the reactive atom is identified by element and atom number. You can pick the reactive atom in the Workspace by clicking the Pick in WS button in the Pick Atom column, and picking an atom in the structure in the Workspace.
- Adsorption types options
-
Choose the types of adsorption sites to consider in the enumeration. At least one type must be selected.
-
Top— Include adsorption sites which are centered on single atoms. The adsorbate atom is bonded to substrate atoms which have been selected as potential adsorption sites by a zero-order bond in the complex.
-
Bridge— Include adsorption sites formed from two substrate atoms which have been selected as potential adsorption sites. Sites are centered between pairs of substrate atoms within (a + b) * X of each other, where a is the covalent diameter of the adsorbate atom, b is the sum of the covalent radii of the two substrate atoms, and X is the neighbor factor defined in the Neighbor factor text box. The adsorbate molecule is bonded to the site by a zero-order bond in the complex. Only available if Include surface atoms within option is selected.
-
Hollow— Include adsorption sites formed from three substrate atoms which have been selected as potential adsorption sites. Sites are centered between three atoms, where each atom is within (a + b) * X of another. a is the covalent diameter of the adsorbate atom, b is the sum of the covalent radii of the two substrate atoms, and X is the neighbor factor defined in the Neighbor factor text box. The adsorbate molecule is bonded to the site by a zero-order bond in the complex. Only available if Include surface atoms within option is selected.
-
- Neighbor factor text box
-
Affects the distance used to consider which substrate atoms can form bridge or hollow adsorption sites. See Bridge or Hollow for information on how this factor is used.
Only available when Bridge or Hollow is selected as the adsorption type.
- Adsorption direction options
-
Choose an option for the direction in which the adsorbate approaches the substrate. The direction is defined by the line (or bond) between the adsorbate reactive atom and the substrate adsorption site (for a given choice of these two in the enumeration).
-
Automatic—use the direction choice (from the two that follow) that results in the shortest bond to the substrate without steric clashes.
-
Along c vector or Z-axis—the adsorbate approaches in the direction of the c vector for a periodic system or the Z axis for a nonperiodic system (cluster).
-
Coordination gap—the adsorbate approaches in the direction of a gap in the substrate site's coordination.
-
- Enumerate N rotations around the adsorption axis option and text box
-
Enumerate the orientations of each adsorbate by rotating it around the adsorption direction, to produce the number of orientations specified in the text box. The adsorbate is rotated by the golden angle (137.5°), chosen to avoid symmetries and duplicates. Each orientation of the adsorbate is placed separately, taking into account steric interactions, so the adsorbate orientations on the surface are not necessarily related by a simple rotation.
Only available when the Molecular option is selected in the Gas molecules section.
- Create additional pre-adsorption structure option
-
If this option is selected, a pre-adsorption complex is created for each generated structure and added to the Entry List under the pre-adsorption subgroup. For systems with periodic boundary conditions, the gas molecule of the pre-adsorption complex is displaced vertically to be halfway in the vacuum gap. For systems without periodic boundary conditions, the gas-substrate bond is lengthened by 5.0 angstroms. Structures that contain overlapping atoms are placed in the Bad Contacts subgroup within the pre-adsorption subgroup.
Only available when the Molecular option is selected in the Gas molecules section.
- Remove symmetry equivalent structures option
-
Remove symmetry equivalent structures in the enumerated set for substrates with periodic boundary conditions. This is helpful to decrease the number of structures for further calculations, such as periodic DFT calculations with Quantum ESPRESSO, or calculation of adsorption energies with the Adsorption Energy Calculations Panel.
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Enumerate Adsorbates - Job Settings Dialog Box, where you can make settings for running the job.
- Status bar
-
The status bar displays information about the current job settings and status for the panel. The settings includes the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
Use the Reset button
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel. The status bar also contains the Help button
 , which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a
, which opens the help topic for the panel in your browser. If the panel is used by one or more tutorials, hovering over the Help button displays a  button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.
button, which you can click to display a list of tutorials (or you can right-click the Help button instead). Choosing a tutorial opens the tutorial topic.