Coarse-Grained Force Field Builder Panel
Build a coarse-grained force field for dissipative particle dynamics (DPD) or Martini modeling from an all-atom system, by fitting parameters for coarse-grained simulations to reproduce all-atom simulations.
Outputs of the Coarse-Grained Mapping Panel can be used in this panel for force field fitting.
The force field can then be used in the Coarse-Grained Force Field Assignment Panel to set up a system for simulation with the MD Multistage Workflow Panel.
To open this panel: click the Tasks button and browse to Materials → Classical Mechanics → Coarse Grain Models → Coarse-Grained Force Field Builder.
The following licenses are required to use this panel: MS Maestro, MS CG, OPLS (optional), MS Force Field Applications (optional), Desmond
- Overview
- Features
- Additional Resources
Overview of the Coarse-Grained Force Field Builder
The all-atom-system is built using the technology of the Disordered System Builder Panel. When the atoms are mapped to particles, a coarse-grained system is constructed from the all-atom system as a reference for comparison with the predictions of the fitted force field for the coarse-grained system. The fitting is done by comparing the distribution of bond lengths and bond angles between the reference and the coarse-grained simulation, and for nonbonded interaction, the radial distribution functions are compared.
Outputs of the Coarse-Grained Mapping Panel can be used in this panel for force field fitting using the Use existing CG-mapped system option.
Particles are defined in terms of SMARTS patterns, and can consist of molecule fragments, a single molecule, or multiple molecules. For multiple molecules, the SMARTS pattern must match whole molecules.
To visualize the results of the calculation and analyze the quality of the fitting parameters, you can use the Coarse-Grained Force Field Viewer Panel (click the Tasks button and browse to Materials → Classical Mechanics → Coarse Grain Models → Coarse-Grained Force Field Builder Results).
To open this panel from the entry group for the results of a job .
.
Coarse-Grained Force Field Builder Panel Features
- Load Selected Entries button
- Coarse-graining type option
- Use force field option and menu
- Import New button
- Atomistic Input tab
- Map Atoms tab
- Mapped Atoms tab
- Use automated CG mapping option
- Target particle size N atoms text box
- Initial Auto Map button
- Particle mapping tools
- Add SMARTS Group button
- Delete All button
- Add SMARTS from FF button
- Allow Overlapping In All button
- Redo Auto Map button
- Allow unique bonds option
- Use particle names from FF option
- Treat small ions implicitly option
- Allow particles to belong to multiple repeating units option
- Add antifreeze water molecules option
- Stabilize ring conformations option
- Estimate Volume from Structure button
- Show Mapped Molecules button
- FF Parameters tab
- CG Simulation tab
- Fitting tab
- Job toolbar
- Status bar
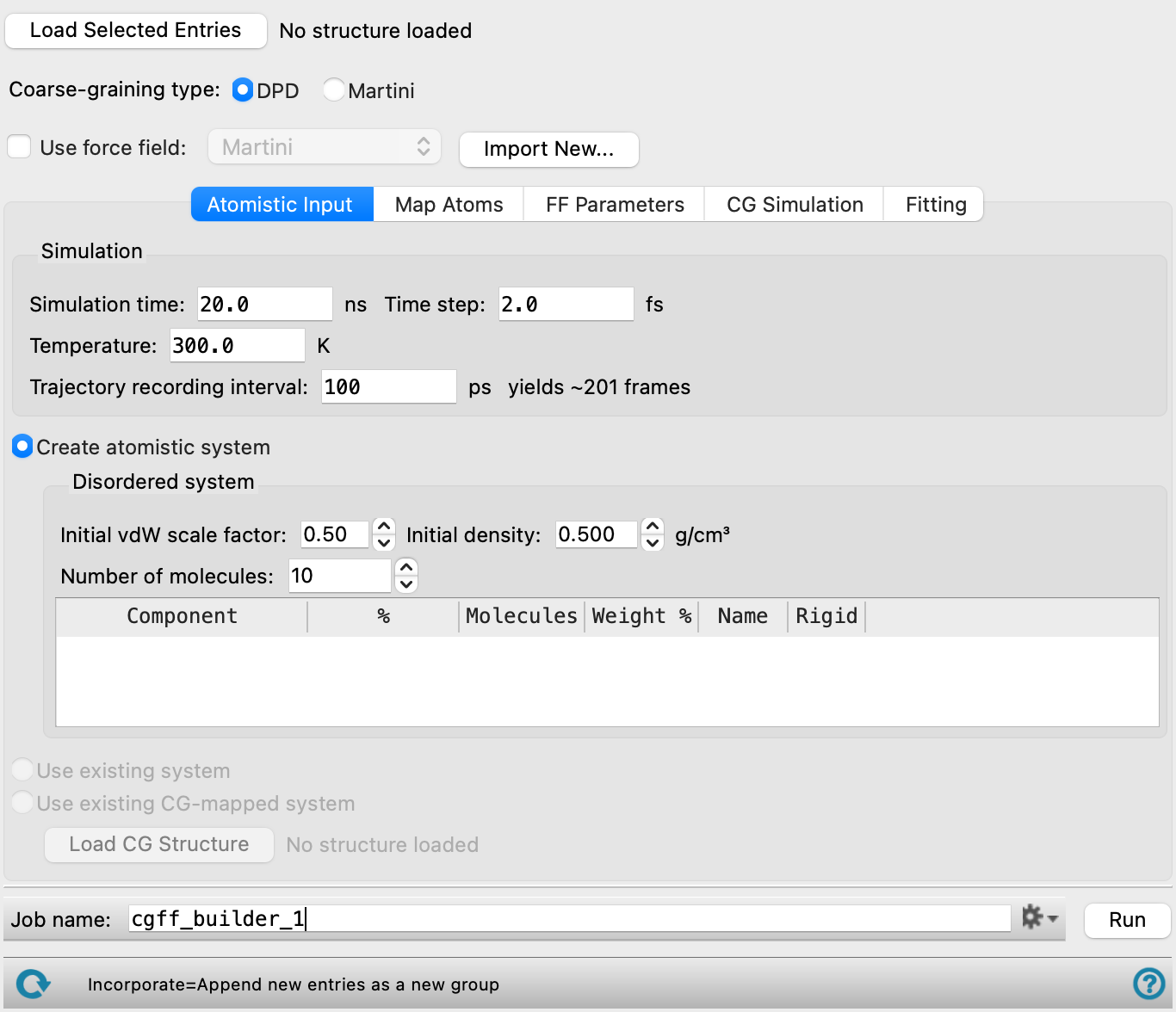
- Load Selected Entries button
-
Load the entries that are selected in the Entry List or Project Table for (1) the all-atom molecules used to build a disordered system or (2) the all-atom system output from the Coarse-Grained Mapping Panel. The structure name and whether of not it contains any trajectory or energy data is displayed to the right of the button.
- Coarse-graining type option
-
Specify the type of coarse-graining force field to build between dissipative particle dynamics (DPD) or Martini modeling.
- Use force field option and menu
-
Use parameters from an existing coarse-grained force field that is stored in your user resources directory. Select the force field from the menu. When the disordered structure is analyzed and the mapping to particles is set up, the parameters from this force field are used to initialize any parameters in the new coarse-grained system, and are fixed at these values by default. When using the Martini_solution force field from this option menu or a custom force field that includes Martini_solution, the bond, angle, and nonbonded parameters are encrypted.
- Import New button
-
Import a force field from a previous run of the coarse-grained force field builder. This force field is added to the Use force field menu, and is also added to your Schrödinger user resources directory. You should therefore make sure that any force field you import is one that you want to keep.
- Atomistic Input tab
-
Define the all-atom system on which the coarse-grained system is based.
- Simulation section
-
Specify the parameters for the all-atom simulation. The disordered system is equilibrated using the default materials relaxation protocol (see Relaxation protocol option and menu), followed by a 1 ns simulation at high pressure and a 1 ns at atmospheric pressure in the NPT ensemble (both at the specified temperature). The production simulation is then run in the NVT ensemble with the parameters specified here.
- Simulation time text box
-
Specify the desired simulation time in ps.
- Time step text box
-
Specify the time step for the simulation in fs.
- Trajectory recording interval text box
-
Set the recording interval for saving points on the trajectory, in ps. This is the amount of time between frames in the trajectory. The entered value is rounded to an integer multiple of the far time step size. The resultant number of records to be written is reported to the right.
- Temperature text box
-
Specify the temperature to be used, in kelvin.
- Create atomistic system option
-
Select this option to create an all-atom system with the parameters specified below.
The system is constructed as a disordered system (see the Disordered System Builder Panel for more information) and equilibrated, then a production simulation is run as a reference for the coarse-grained simulations.
- Disordered system section
-
Specify the composition of the disordered system and parameters for constructing it. The components are placed using the tangled chain algorithm (see Tangled chain option).
- Initial vdW scale factor text box
-
Specify a scaling factor for the van der Waals radii of the atoms, to determine when atoms clash. The scaling factor is adjusted if necessary, to avoid clashes. Atoms are considered hard spheres and any overlap is considered a steric clash. The scaling factor is used in the optimization of the packing of the components. A smaller factor allows closer packing but also allows more clashes.
- Initial density text box
-
Specify the initial density of the system in g cm-3. The density may change during the equilibration.
- Number of molecules text box
-
Specify the total number of molecules in the disordered system.
- Component table
-
Specify the composition of the disordered system. The components are the molecules loaded using the Load Selected Entries button, and are listed by entry title. You can specify the composition as a percentage by number, a number of molecules, or a percentage by weight. Editing one of these values in the table automatically updates the others. The values are adjusted to ensure that the specified total number of molecules is obtained.
- Use existing system option
-
Use an existing all-atom disordered system for assignment of coarse-grained particles. The existing input simulation should contain cms, trajectory, and ene files.
- Use existing CG-mapped system option
-
Use an existing coarse-grained mapped system for building a force field. Only available when the loaded structure is an output of the Coarse-Grained Mapping Panel.
- Load CG Structure button
-
Load the coarse-grained structure corresponding to the loaded all-atom input. This coarse-grained structure must be output using the Export CG structure to Workspace button in the Mapped Structures tab of the Coarse-Grained Mapping Panel. The file name is displayed to the right after loading.
- Map Atoms tab
- Mapped Atoms tab
-
Map atoms to particles, using SMARTS patterns. This can be done manually or by using automated coarse-grained mapping. Particles can be composed of a group of atoms within a molecule, or they can be composed of multiple entire molecules, e. g. a cluster of water molecules. This means that the SMARTS pattern for a multi-molecule particle must be a pattern for an entire molecule.
When the atoms have been mapped to particles, a coarse-grained system is constructed from the all-atom system. This system serves as a reference for the adjustment of the coarse-grained force field.
Each particle is defined with a set of common tools; you can add a new set of tools for each particle you want to define. These tools are described below.
The order in which you define the particles matters: particles with more specific SMARTS patterns must be defined before particles with less specific patterns (i,e, be higher up in the list of particle mappings). For example, if one particle maps to CCO and another maps to CC, you must define the CCO particle first, otherwise the matches to CC will also match CCO and no particles will be found for CCO. If this is not the case, you can use the management buttons to move the particle definitions up or down the list so that they are in the desired order.
When the Use existing CG-mapped system option is selected, the Mapped Atoms tab displays the noneditable mapping output as determined by the Coarse-Grained Mapping Panel. Many features listed below are not available when in the Mapped Atoms mode.
For proteins mapped using the Martini Coarse-graining type, harmonic restraints are automatically applied to their backbones.
- Use automated CG mapping option
-
Select this option to map atoms to particles automatically given an all-atom system and a target coarse-grained particle size. If selected, the mapping can be fully automated or can be a mixture of manually and automatically defined particles depending on the options specified.
- Target particle size N atoms text box
-
Specify the target number of heavy atoms per coarse-grained particle to use when mapping an atomistic structure to a coarse-grained structure. The panel attempts to map particles with the target number of atoms while honoring the chemistry of the all-atom structure. Some particles may have more or less atoms than the specified target. The specified target is set to 4 for the Martini Coarse-graining type and is noneditable. Only available when the Coarse-graining type is set to DPD and Use automated CG mapping is selected.
- Initial Auto Map button
-
Click this button to automatically map particles taking into account the target particle size. The corresponding SMARTS tool sets are populated and display green text stating Auto-mapped. Only available when Use automated CG mapping is selected.
- Particle mapping tools
-
These tools are used to map a group of atoms to a particle.
- SMARTS tool set management buttons
-
These buttons perform display and ordering operations on the SMARTS tool set. They allow for easy duplication and rearrangement of SMARTS tool sets.


Show or hide the contents of the SMARTS tool set. When hidden, only the SMARTS tool set number, label (if any) and these buttons are displayed. This is useful when you have a number of SMARTS tool sets and want to compare two separate SMARTS tool sets, for example. 

Move the SMARTS tool set up or down one place in the list. 
Duplicate the SMARTS tool set. This is useful for creating similar SMARTS tool sets with variations on the settings. 
Delete the SMARTS tool set. - Particle name text box
-
Specify a name for the particle. The same name can be applied to multiple SMARTS patterns. Particles that are automatically mapped are named automatically and these names are noneditable.
- SMARTS text box and
-
Enter the SMARTS pattern for the atoms in the particle in the text box, or select the atoms in the Workspace for the SMARTS pattern and click
- Map N molecules to 1 particle box
-
Specify the number of molecules to be mapped to this particle. If the number is greater than 1, the SMARTS pattern must specify an entire molecule: a particle cannot be composed of fragments from different molecules. If there is only one molecule mapped to the particle, the SMARTS pattern can be a pattern for part of the molecule, and you can create multiple particles from a single molecule.
-
This parameter is auto-populated when using automated CG mapping.
- Particle volume text box
-
Specify the volume of the particle in Å3. A default value based on the SMARTS pattern provided. Only present when the Coarse-graining type is set to DPD.
- Allow atoms to belong to multiple particles option
-
This option relaxes the restriction that atoms must belong to only the particles of the current SMARTS pattern. Particles can then be connected where there is an atom rather than where there is a bond, which can assist in obtaining particles that are symmetric where otherwise they would be asymmetric (e.g in polypropylene). This is always selected for automatically mapped particles.
Only available when Use automated CG mapping is not selected.
- Use as pre-defined pattern when auto-mapping option
-
Enforce a user-specified mapping of a SMARTS pattern to a particle. The SMARTS set is converted from an automatically mapped set to a user-defined set. Sets with this option selected are mapped first. Any atoms which are not in the user-defined mapping are automatically mapped.
This option is helpful when there are specific mappings that need to be enforced.
Only present when Use automated CG mapping is selected.
- Add SMARTS Group button
-
Add a set of tools for defining a particle, below the existing sets.
- Delete All button
-
Remove all sets of tools defining particles.
- Add SMARTS from FF button
-
Import a predefined SMARTS pattern from the force field selected in the Use force field option menu. The Import Smarts from Force Field dialog opens. Only the most specific SMARTS pattern is shown in the Structure column, but you can hover over the structure to see the other SMARTS patterns associated with the Particle Name. Each associated SMARTS pattern imports as a separate SMARTS group. Only the Particle name and SMARTS pattern are imported, and other mapping parameters in the SMARTS group still need to be defined.
Only available if the Use force field option is selected.
- Allow Overlapping In All button
-
Checks the Allow atoms to belong to multiple particles option for all SMARTS groups in the panel. This is always selected for automatically mapped particles.
Only available when Use automated CG mapping is not selected.
- Redo Auto Map button
-
Click this button to update the automated coarse-grained mapping after making a change to the initial mapping. The text of this button is green when a change that requires redoing the automated mapping is made. Only available when Use automated CG mapping is selected.
- Allow unique bonds option
-
If different types of all-atom bonds map to the same type of coarse-grained bond, this option ensures that distinct coarse-grained bonds are generated for each type of all-atom bond, i.e. a bond parameter is introduced for each unique bond type. Setting this option increases the number of fitting parameters.
- Use particle names from FF option
-
Use the particle names found in the force field file for particles with a matching SMARTS pattern.
-
Only available if the Use force field option is selected.
- Treat small ions implicitly option
-
Small ions, defined as charged molecules with number of heavy atoms less than or equal to the Target mapping size divided by two, are treated implicitly. Coarse-grained particles are not mapped for small ions but the ions are considered in the reference all-atom simulations. Only present when the Coarse-graining type is set to DPD and Use automated CG mapping is selected.
- Allow particles to belong to multiple repeating units option
-
Allow particles on two adjacent repeating units to share atoms. Only present when the Coarse-graining type is set to DPD and Use automated CG mapping is selected.
- Add antifreeze water molecules option
-
Replace 10% of water molecules, (W), with antifreeze molecules, (WF). Only present when the Coarse-graining type is set to Martini. WF particles are identical to W particles, except that they interact with W particles differently, which can reduce the risk for Martini water freezing.
- Stabilize ring conformations option
-
Add diagonal bonds to rings of 4 particles. This is useful for stabilizing the conformation of rigid and planar rings. Only present when the Coarse-graining type is set to Martini and Use automated CG mapping is selected.
- Estimate Volume from Structure button
-
Calculate the particle volume using the structure instead of the SMARTS pattern. Changes the value in the Particle volume text box. This is recommended if Allow atoms to belong to multiple particles is selected for any particle, and if Set using particle volume and charge is selected in the Nonbonded tab of the FF Parameters tab. Only present when the Coarse-graining type is set to DPD.
- Show Mapped Molecules button
-
Click this button to display the coarse-grained structure in the Workspace. A dialog box opens with a Particles section, consisting of particle names, number of occurrences of particles, and their corresponding colors in the Workspace, and a Show section, with the options of visualizing the coarse-grained structure or the mapping superimposed on the all-atom structure. Only unique molecules, including those of different stereochemistries, in the input structure are mapped and displayed. The entry can be saved to the Entry List by clicking the Save Structure button. Clicking Cancel will remove the entry from the Entry List and return to the main panel.
- FF Parameters tab
-
Specify the parameters that are to be included in the coarse-grained simulation, provide initial values, and fix the values of parameters that do not need to be adjusted. For more information on the parameters, see the Coarse-Grained Force Field Assignment Panel topic.
- Populate Using Structure button
-
Populate the tables for the different parameter types from the all-atom disordered system structure, and fill in the initial values. This is required for angle parameters. If you run a job without populating the tables, the force field will not have angle parameters, but the other parameters will be fitted (non-bonded force constants, bond distances, and bond force constants).
- Import from Force Field button
-
Import the parameters from the force field chosen from the Use force field option menu. When using the Martini_solution force field or a custom force field that includes Martini_solution, the bond, angle, and nonbonded parameters are encrypted.
- Particle tab
-
Specify charges for the particles, if necessary.
- Use common mass option and text box
-
Specify a mass in amu to use for all particles. If this option is not selected, mass is automatically determined. Only present when the Coarse-graining type is set to DPD.
- Charge type options
-
Select Implicit or Explicit to define the charges on the particle. Only present when the Coarse-graining type is set to DPD. For the Martini Coarse-graining type, charges are explicit.
- Particle type table
-
For each particle type, specify the charge. Values can only be -1, 0, or 1. You can sort the particle types by clicking the arrow in the heading cell.
For the Martini Coarse-graining type, the Martini type option menu is present and set to Custom for each particle. If a Martini type other than Custom is specified by the user, the values of the nonbonded parameters, Lennard-Jones interaction strength (ε/(kcal mol-1)) and range (σ/Å), are fixed for (1) self interactions of non-custom particles and (2) interactions between two non-custom particles. For interactions including one or two custom particles, the usual nonbonded parameter fitting options are available. See https://cgmartini.nl/docs/downloads/force-field-parameters/martini2/particle-definitions.html or Reference 15 for more information on Martini types.
- Bond tab
-
Specify whether to fix bond distance and force constant for bonds between particles.
- Bond type table
-
For each bond, specify how to treat the bond distance (Req/Å) and force constant for bonds between particles (k/(kcal mol-1 Å-2)):
- Fit—the parameter is included in the coarse-grained simulation and optimized throughout the course of the simulation.
- Initialize—a text box appears to specify a value, which is used in the first step of the simulation. The unit for the value is given in the column header after the slash (/). The value is then optimized throughout the course of the simulation.
- Fixed—a text box appears to specify a value, which is fixed during the simulations. The unit for the value is given in the column header after the slash (/).
- Angle tab
-
Set the initial angles and force constants for the angles between particle triads. For the Martini Coarse-graining type, any angle potentials for which the three particles constituting the angle are also bonded in a triangle in the coarse grained structure are excluded from the fitting.
- Ignore All Angles button
-
Ignore angles in the simulation.
- Angle settings table
-
For each angle, specify how to treat the equilibrium angle (θeq/°) and force constant for angles (k/(kcal mol-1 rad-2) or k/(kcal mol-1)). You can delete specific angles by clicking the icon in the Delete column.
- Fit—the parameter is included in the coarse-grained simulation and optimized throughout the course of the simulation.
- Initialize—a text box appears to specify a value, which is used in the first step of the simulation. The unit for the value is given in the column header after the slash (/). The value is then optimized throughout the course of the simulation.
- Fixed—a text box appears to specify a value, which is fixed during the simulations. The unit for the value is given in the column header after the slash (/).
- Nonbonded tab
-
Specify parameters for nonbonded interactions.
- Dielectric constant text box
-
Set the dielectric constant for the system. If water is modeled as a sphere, for example, it has no dipole moment, so a dielectric constant is needed to model the interaction with the water.
- Cutoff distance text box
-
Specify the maximum distance between particles for including a nonbonded interaction. Only available when the Coarse-graining type is set to DPD.
- Reduced density text box
-
Specify the reduced density. Only present when the Coarse-graining type is set to DPD.
- Nonbonded parameter table
-
For each nonbonded interaction, specify how to treat the repulsive harmonic potential (a/(kcal mol-1 Å-2)) for the DPD Coarse-graining type and the Lennard-Jones interaction strength (ε/(kcal mol-1)) and range (σ/Å) for the Martini Coarse-graining type.
- Fit—the parameter is included in the coarse-grained simulation and optimized throughout the course of the simulation.
- Initialize—a text box appears to specify a value, which is used in the first step of the simulation. The unit for the value is given in the column header after the slash (/). The value is then optimized throughout the course of the simulation.
- Fixed—a text box appears to specify a value, which is fixed during the simulations. The unit for the value is given in the column header after the slash (/).
- Set using particle volume and charge—values are calculated once and then fixed. This last option should only be used for interactions between particles of the same type. Only present when the Coarse-graining type is set to DPD.
- CG Simulation tab
-
Specify the parameters for the coarse-grained production simulations used for fitting the force-field parameters.
The simulation first performs a Brownie minimization, followed by simulations of 100ps at 10K and 1ns at 300K in the NVT ensemble, followed by the production simulation in the NVT ensemble.
- Ensemble class option menu
-
Choose the ensemble class from this option menu. The following classes are available:
- NVT—constant particle number (N), volume (V) and temperature (T). This class represents the canonical ensemble.
- NPT—constant particle number (N), pressure (P) and temperature (T). This class is an isothermal-isobaric ensemble, the common experimental conditions.
-
Only present when Martini is selected as the Coarse-graining type. For DPD, NVT is always used.
- Simulation time text box
-
Specify the desired simulation time in ps.
- Time step text box
-
Specify the time step for the simulation in fs.
- Temperature text box
-
Specify the temperature to be used, in kelvin.
- Trajectory recording interval text box
-
Set the recording interval for saving points on the trajectory, in ps. This is the amount of time between frames in the trajectory. The entered value is rounded to an integer multiple of the far time step size. The resultant number of records to be written is reported to the right.
- Fitting tab
-
Specify parameters for fitting the force field from the coarse-grained simulation results.
- Maximum ε optimized per iteration text box
-
Specify the maximum number of epsilon, ε, updates per iteration. Please note that using larger values here may speed up convergence but can also increase instability. Only available when Martini is selected as the Coarse-graining type.
- Analyze last N % of simulation text box
-
Specify the percentage of the production coarse-grained simulation to use for the fitting of the force field parameters, taken from the end of the simulation.
- Distance resolution text box
-
Specify the resolution for the calculation of the radial distribution function and the binning of the bond distances. The bond length distribution and the radial distribution function are used to compare the coarse-grained simulation results with the reference.
- Iterations text box
-
Set the number of iteration to use for fitting the parameters. Each iteration consists of a coarse-grained simulation with a set of parameters, then an adjustment of the parameters to improve the reproduction of the results of the reference all-atom simulation.
- Job toolbar
-
Manage job submission and settings. See Job Toolbar for a description of this toolbar.
The Job Settings button opens the Coarse-Grained Force Field Builder - Job Settings Dialog Box, where you can make settings for running the job.
- Status bar
-
 to reset the panel to its default settings and clear any data from the panel.
to reset the panel to its default settings and clear any data from the panel.If you can submit a job from the panel, the status bar displays information about the current job settings and status for the panel. The settings include the job name, task name and task settings (if any), number of subjobs (if any) and the host name and job incorporation setting. The job status can include messages about job start, job completion and incorporation.
The status bar also contains the Help button
 , which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.
, which opens an option menu with choices to open the help topic for the panel (Documentation), launch Maestro Assistant, or if available, choose from an option menu of Tutorials. If the panel is used by one or more tutorials, hover over the Tutorials option to display a list of tutorials. Choosing a tutorial opens the tutorial topic.